NHRI Communications

知識產權
新藥大放異彩-漫談2014年FDA核准小分子新藥(中)「註1」
Introduction to FDA approved novel small molecular drugs in 2014 (part 2)
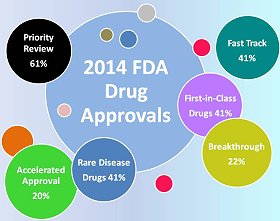 上集提到2014年是自1996年以來新藥睽違已久的豐收年,獲准新藥數目最多的一年,共有41個,其中以癌症藥物與感染性疾病藥物為主要領域。2014年共有8個癌症治療藥物與1個癌症化療止吐藥獲准上市(占所有核准新藥的22%),在感染性疾病藥物方面,除了2個抗C型肝炎藥物外,尚有9個藥物核准用於治療細菌、真菌、流感病毒及利什曼原蟲所引起的感染疾病。相較於2012年與2013年,癌症藥物維持一定的比率;但感染性疾病用藥卻從2012年4個及2013年3個,大幅增長到2014年的11個,其中有4個是被授予合格感染性疾病產品資格(Qualified Infectious Disease Product, QIDP)而獲得FDA核准的抗生素(如表一所示)。
上集提到2014年是自1996年以來新藥睽違已久的豐收年,獲准新藥數目最多的一年,共有41個,其中以癌症藥物與感染性疾病藥物為主要領域。2014年共有8個癌症治療藥物與1個癌症化療止吐藥獲准上市(占所有核准新藥的22%),在感染性疾病藥物方面,除了2個抗C型肝炎藥物外,尚有9個藥物核准用於治療細菌、真菌、流感病毒及利什曼原蟲所引起的感染疾病。相較於2012年與2013年,癌症藥物維持一定的比率;但感染性疾病用藥卻從2012年4個及2013年3個,大幅增長到2014年的11個,其中有4個是被授予合格感染性疾病產品資格(Qualified Infectious Disease Product, QIDP)而獲得FDA核准的抗生素(如表一所示)。
感染性疾病用藥
1928年亞歷山大.弗萊明(Alexander Fleming)發現青黴素(penicillin),被視為現代製藥工業抗生素之濫觴。但在過去十幾年,抗生素領域卻一直是製藥業的冷門領域,主要原因是一般診所使用的抗生素因失去專利保護而售價低廉,而醫院使用的抗生素,多用於急症,用藥時間短,利潤率極低。另外,舊抗生素在治療多數常見感染仍相對有效,使得新型抗生素的研發變得非常尷尬,再加上透過高速藥物篩選衍生的藥物,常因體外活性很好但體內療效多數不盡人意,而難以與天然產物相比,同時新型抗生素的開發也很繁瑣,適應症小而雜。再者,抗生素用藥時間短,所以單價必須很高才能盈利,這使得很多大型藥廠基於獲利偏低的考量,紛紛削減了抗生素領域的投資。
最近,美國疾病控制和預防中心(Centers for Disease Control and Prevention, CDC)研究報告指出,僅在美國每年就有超過200萬人因為感染抗藥性病菌而患病,其中至少2.3萬人因此喪生。英國廣播公司(BBC)亦報導,預估到2050年全球每年將有1,000萬人死於抗藥性病菌感染,數量將超過目前癌症的死亡人數。目前全球每年約有70萬人死於抗藥性病菌感染,故全世界亟需針對抗藥性病菌進行預防與治療,以解決日益嚴峻的抗藥性病菌傳播問題;因此,美國政府及國會對於具強烈抗藥性的病原體、致命醫療醫院內的集體細菌感染與潛在對於公共衛生的嚴重威脅等抗藥性病菌所引發的傳染性疾病高度重視,尤其是「抗藥性金黃色葡萄球菌(Methicillin-resistant Staphylococcus aureus或multiple-resistant Staphylococcus aureus, MRSA)」對於最後一道防線抗生素的抗藥性日漸增加之威脅。因此,美國總統歐巴馬在2012年7月簽署通過《鼓勵開發抗生素法案(Generating Antibiotic Incentives Now (GAIN) Act)》,其中明定獎勵新開發的抗生素條款,例如符合《GAIN法案》之抗生物藥物,新增額外5年的市場獨占排他權,且承諾在接受申請後的6個月內必須啟動審評,並且在審查和跟蹤進度時亦享有優先權。此外,該法案還為部分符合條件的抗生素設立「合格感染性疾病產品」分類,對於能治療特定難治的病原體的藥物,例如治療抗藥性金黃色葡萄球菌的抗生素,還能獲得額外的5年獨占排他權。總之,適用於《GAIN法案》的「合格感染性疾病產品」若為新藥,則會享有至少9年的非專利性市場排他銷售期;若是舊藥新用則有8年;而用於罕見疾病的合格感染性疾病產品則有12年的保護。有別於由專利批准所帶來的獨占排他權,此處所提之市場獨占排他權是由FDA賦予某特定藥品的獨占期間,即使獲得獨占權的藥品本身並沒有獲得專利排他權,FDA也不會核准同類藥品上市。
由於美國政府帶頭祭出鼓勵抗生素研發法案,並且設立新的獎勵措施,使得包括羅氏(Roche)、葛蘭素史克(GSK)等大型藥廠近期已紛紛投資,甚至啟動新一輪的藥廠併購戲碼,例如阿特維斯(Actavis)以6.75億美元收購Durata Therapeutics公司、默克(Merck)斥資95億美元取得Cubist公司,進而讓大藥廠重新加入抗生素藥物的競技場,以迎接「後抗生素時代」到來。以下是2014年9個感染症藥物(Dalvance®、Sivextro®、OrbactivTM、ZerbaxaTM、Jublia®、KerydinTM、XtoroTM、RapivabTM、Impavido®)之相關開發過程:
(1) Dalvance® (dalbavancin)
 2014年5月23日Dalvance®(dalbavancin)拔得頭籌,成為2014年第1個獲得FDA以「合格感染性疾病產品」資格批准的抗生素,主要是治療成人由細菌,如金黃色葡萄球菌(包括MRSA)、化膿性鏈球菌所引起的急性皮膚及皮膚組織感染(acute bacterial skin and skin structure infections, ABSSSI)。Dalvance®總共進行21項臨床試驗,其中包括約有3,000例患者參與之5個臨床III期試驗,其治療急性皮膚及皮膚組織感染的效果與萬古黴素(vancomycin)相當;不過,Dalvance®較萬古黴素的活性來得強(只需2週療程)且具較長半衰期(1週1次靜脈注射vs.萬古黴素需每天多次靜脈注射),使得Dalvance®成為me-better藥物(類新藥,增進現有產品效果及減低副作用)。Dalvance®屬於第二代半合成的脂糖肽類抗生素,主要是針對萬古黴素和teicoplanin抗生素之糖肽主結構進行親脂性側鏈修飾。
2014年5月23日Dalvance®(dalbavancin)拔得頭籌,成為2014年第1個獲得FDA以「合格感染性疾病產品」資格批准的抗生素,主要是治療成人由細菌,如金黃色葡萄球菌(包括MRSA)、化膿性鏈球菌所引起的急性皮膚及皮膚組織感染(acute bacterial skin and skin structure infections, ABSSSI)。Dalvance®總共進行21項臨床試驗,其中包括約有3,000例患者參與之5個臨床III期試驗,其治療急性皮膚及皮膚組織感染的效果與萬古黴素(vancomycin)相當;不過,Dalvance®較萬古黴素的活性來得強(只需2週療程)且具較長半衰期(1週1次靜脈注射vs.萬古黴素需每天多次靜脈注射),使得Dalvance®成為me-better藥物(類新藥,增進現有產品效果及減低副作用)。Dalvance®屬於第二代半合成的脂糖肽類抗生素,主要是針對萬古黴素和teicoplanin抗生素之糖肽主結構進行親脂性側鏈修飾。Dalbavancin原屬於Vicuron Pharmaceuticals公司的開發藥品,早在2004年12月即向FDA提出新藥查驗登記(New Drug Application, NDA),爾後輝瑞藥廠(Pfizer)在2005年6月以19億美元併購,旋即取得Vicuron Pharmaceuticals之兩項臨床III期產品anidulafungin和dalbavancin。2007年底dalbavancin獲FDA有條件上市核可,但需要提供進一步的臨床相關試驗資料;Pfizer於是在2008年9月宣布撤銷dalbavancin之NDA,並重啟另一個臨床III期試驗。不久卻由在2009年12月才成立於美國芝加哥的新創公司Durata Therapeutics,僅以1,000萬美元即全盤接手dalbavancin之後續開發權利,且Pfizer仍需繼續支付額外600萬美元用於臨床試驗費用。二者協議,如果在5年內dalbavancin獲准在美國或歐洲上市,Durata Therapeutics需支付輝瑞藥廠2,500萬美元作為補償。之後,Durata Therapeutics在2011年4月另啟一個Dalvance®臨床III期試驗(DISCOVER-1),最終在2014年獲得FDA核可上市。市場預估Dalvance®到2019年將可有4.5億美元之銷售額,因此引起總部位於愛爾蘭都柏林的Actavis公司(前身為美國Watson製藥,創辦人為趙宇天博士)之注意,於2014年10月6日以6.75億美元併購Durata Therapeutics,主要是看中治療皮膚感染的抗生素藥品Dalvance®之未來潛力。JP Morgan分析,到2020年Dalvance®在美國市場可達到2.04億美元銷售額。
(2) Sivextro® (tedizolid phosphate)
 Sivextro® (tedizolid phosphate, torezolid phosphate, TR-701, DA-7218)主要是治療成人由細菌,如金黃色葡萄球菌(包括MRSA)、各種鏈球菌或腸球菌所引起的急性皮膚及皮膚組織感染,其結構屬於oxazolidinone類抗生素之前驅藥(prodrug),在體內可被磷酸酶迅速轉換成原藥tedizolid,並與細菌的核醣體50S亞基結合,進而抑制細菌蛋白質之合成。雖然自2000年Pfizer的oxazolidinone類抗生素Zyvox®(linezolid)核准上市後,至少有10個類新藥(me-too)進入臨床試驗,但是Sivextro®是第一個獲得FDA核准的第二代oxazolidinone類抗生素。相較之下,Sivextro®對於一些細菌的體外抑制活性比Zyvox®來得強2-8倍,安全性亦有所提高,所需劑量也從1天2次600毫克Zyvox®大幅減少到1天1次200毫克Sivextro®,且療程也從10天縮短到6天,可謂是從me-better藥物到最佳(best-in-class)藥物的代表。由於Sivextro®是用於治療嚴重或危及生命的皮膚及皮膚組織感染,故於2013年12月13日獲得FDA的優先審查資格,且因符合「合格感染性疾病產品」上市資格,獲得延長5年的市場獨占權,於2014年6月20日獲准上市。JP Morgan分析,到2020年Sivextro®在美國市場可達到2.16億美元銷售額。
Sivextro® (tedizolid phosphate, torezolid phosphate, TR-701, DA-7218)主要是治療成人由細菌,如金黃色葡萄球菌(包括MRSA)、各種鏈球菌或腸球菌所引起的急性皮膚及皮膚組織感染,其結構屬於oxazolidinone類抗生素之前驅藥(prodrug),在體內可被磷酸酶迅速轉換成原藥tedizolid,並與細菌的核醣體50S亞基結合,進而抑制細菌蛋白質之合成。雖然自2000年Pfizer的oxazolidinone類抗生素Zyvox®(linezolid)核准上市後,至少有10個類新藥(me-too)進入臨床試驗,但是Sivextro®是第一個獲得FDA核准的第二代oxazolidinone類抗生素。相較之下,Sivextro®對於一些細菌的體外抑制活性比Zyvox®來得強2-8倍,安全性亦有所提高,所需劑量也從1天2次600毫克Zyvox®大幅減少到1天1次200毫克Sivextro®,且療程也從10天縮短到6天,可謂是從me-better藥物到最佳(best-in-class)藥物的代表。由於Sivextro®是用於治療嚴重或危及生命的皮膚及皮膚組織感染,故於2013年12月13日獲得FDA的優先審查資格,且因符合「合格感染性疾病產品」上市資格,獲得延長5年的市場獨占權,於2014年6月20日獲准上市。JP Morgan分析,到2020年Sivextro®在美國市場可達到2.16億美元銷售額。值得一提的是,Cubist公司並不是Sivextro®原始開發商,而是透過併購而取得Sivextro®之所有權。Cubist於2013年7月31日分别以5.35億美元和7.07億美元收購位於美國聖地牙哥的Optimer和Trius2家藥廠。收購Optimer藥廠主要是針對該公司年銷售6,000萬美元的Dificid®;收購Trius藥廠則是為取得原為TR-701的Sivextro®之所有權。然而,TR-701也不是Trius藥廠自行研發的產品,而是Jeffrey Stein博士在2007年1月創立Trius藥廠(前身為Rx3 Pharmaceuticals)時,於2007年2月27日專門從南韓東亞製藥(Dong-A Pharmaceuticals)取得除南韓和北韓以外之全球授權,故Trius藥廠可謂是Jeffrey Stein博士慧眼看上TR-701之潛力而創辦的新創公司,並於2007年起在美國進行臨床I期試驗,一直到2014年6月20日獲准上市。另一方面,東亞製藥對Trius藥廠授權,可獲得1,700萬美元研發里程碑金,以及上市後12年的權利金收入(5-7%全球銷售額,初估每年2,000萬美元),也使得東亞製藥繼2003年LG生命科學研發的Factive®(gemifloxacin)抗生素之後,成為韓國製藥公司第2次獲得美國FDA核准新藥上市,替南韓新藥研發再添新頁。
(3) OrbactivTM (oritavancin)
 2014年8月6日OrbactivTM(oritavancin, LY333328)成為2014年第3個以「合格感染性疾病產品」通過FDA審核的抗生素,屬於第二代半合成的脂糖肽類抗生素,由天然產物chloroerymomycin所衍生而來,其抗菌活性較萬古黴素高出1,000倍之多。主要是治療成人由細菌,如金黃色葡萄球菌(包括MRSA)、各種鏈球菌或腸球菌所引起之急性皮膚及皮膚組織感染,是治療此類感染抑制劑中唯一單一藥物的抗生素,患者只需要接受1次OrbactivTM靜脈注射,即可完成整個療程。從SOLO I和SOLO II臨床III期研究數據證明,僅注射一次OrbactivTM(1,200毫克,IV)與7-10天每天注射2次萬古黴素(1,000毫克,IV)之療效相當,同時亦比萬古黴素降低19.2%副作用的發生率。
2014年8月6日OrbactivTM(oritavancin, LY333328)成為2014年第3個以「合格感染性疾病產品」通過FDA審核的抗生素,屬於第二代半合成的脂糖肽類抗生素,由天然產物chloroerymomycin所衍生而來,其抗菌活性較萬古黴素高出1,000倍之多。主要是治療成人由細菌,如金黃色葡萄球菌(包括MRSA)、各種鏈球菌或腸球菌所引起之急性皮膚及皮膚組織感染,是治療此類感染抑制劑中唯一單一藥物的抗生素,患者只需要接受1次OrbactivTM靜脈注射,即可完成整個療程。從SOLO I和SOLO II臨床III期研究數據證明,僅注射一次OrbactivTM(1,200毫克,IV)與7-10天每天注射2次萬古黴素(1,000毫克,IV)之療效相當,同時亦比萬古黴素降低19.2%副作用的發生率。OrbactivTM曾幾度易手。OrbactivTM(LY333328)最初是由禮來藥廠(Eli Lilly)研發,但2001年Eli Lilly以5,000萬美元以及階段里程碑金和上市權利金,將OrbactivTM授權給InterMune公司。InterMune為了將產品線聚焦在肝臟和肺臟藥物,故在2005年將OrbactivTM轉讓給Targanta Therapeutics公司,InterMune和Eli Lilly並於2007年分別收到400萬和100萬美元里程碑金。Targanta Therapeutics於2008年向FDA提出NDA,但遭到FDA拒絕,並要求補充申報資料。2009年1月12日,The Medicines Company以4,200萬美元併購Targanta Therapeutics,並取得OrbactivTM後續開發權,在進行多項臨床試驗後,終於在2014年2月再次向FDA提出NDA,並在同年9月6日獲得核准上市。JP Morgan分析,到2020年OrbactivTM在美國市場可達到3.09億美元銷售額。
(4) ZerbaxaTM (ceftolozane/tazobactam)
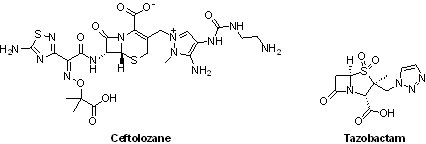
ZerbaxaTM(ceftolozane/tazobactam, CXA-201)是一種複方抗生素,用於治療成人患者因感染革蘭氏陰性菌(Gram-negative bacteria)導致的複雜性尿路感染(cUTI)和複雜性腹腔內感染(cIAI)。主要成分一是ceftolozane(CXA-101, FR-264205),屬於頭孢類抗生素(oxyimino-aminothiazolyl cephalosporin),主要作用為抑制細菌細胞壁之合成;另一個成分tazobactam,屬於β-內醯胺酶抑制劑(β-lactamase inhibitor),用以增加抗生素之廣效性。類似的複方抗生素藥物有Zosyn®(piperacillin/tazobactam)、Augmentin®(amoxicillin/clavulanic acid)、Timentin® (ticarcillin/clavulanic acid)、Unasyn®(ampicillin/sulbactam)與Sulperazon® (cefoperazone/sulbactam)。
Ceftolozane(FR-264205)原屬於日本藤澤藥品(Fujisawa Pharmaceutical),後因在2005年與山之內製藥(Yamanouchi Pharmaceutical)合併組成安斯泰來製藥(Astellas Pharma),故FR-264205所有權改為Astellas Pharma擁有。2007年Eckard Weber和James Ge2位博士在美國聖地牙哥創立Calixa Therapeutics公司,並取得FR-264205(後改名為CXA-101)除亞洲以外之全球開發權,並以CXA-201(ceftolozane/tazobactam)為主要旗艦產品。2009年12月14日Cubist公司以4.02億美元併購Calixa Therapeutics,進而取得ZerbaxaTM之所有權。
ZerbaxaTM是2014年FDA第4個通過符合「合格感染性疾病產品」資格審查上市的抗生素(2014年12月19日),為核准治療革蘭氏陰性菌的首項新抗生素產品。據估計,在美國每年由革蘭氏陰性菌感染導致的病例高達200萬例,死亡病例有2.3萬例,產生的直接醫療費用高達200億美元。RBC Capital Markets預估,ZerbaxaTM未來每年將有15億美元之銷售額,而2015年2月25日FDA通過由Actavis和AstraZeneca共同開發的類新藥AvycazTM(ceftazidime/avibactam),預計將會瓜分此巨大的革蘭氏陰性菌感染藥物之市場。
最後,值得注意的是,Cubist是一家以開發抗生素藥物見長之生技公司,由於過去主流大藥廠視抗生素為夕陽產業,不願意投入研發資金,使得Cubist能夠以每年4億美元的研發預算就成為抗生素研發領域的領頭羊,擁有多項產品上市和臨床開發。尤其是2014年對於Cubist而言,可說是豐收年,除了2003年通過FDA核准上市、年銷售額高達10億美元,用以對抗格蘭氏陽性菌(Gram-positive bacteria)的旗艦藥救必辛Cubicin® (daptomycin)外,2014年同時又有2項抗生素藥物獲得FDA核准上市-Sivextro®和ZerbaxaTM。不過,最近FDA聯邦法院判決Cubicin®之4項專利無效,並裁決Hospira公司最快可在2016年推出Cubicin®仿製藥,比預期專利到期時間提前2年,此專利延伸敗訴將會縮短Cubicin®年度營業額。由於Cubist藥廠之小而美的成功,引起多家主流大藥廠之併購考量,最後由美國Merck於2014年12月9日以84億美元現金收購Cubist,另外亦包括11億美元的債務在內,故整個交易價值高達95億美元,此乃Merck於2014年的第2次重大交易,在此之前,Merck在6月即斥資38.5億美元收購Idenix製藥,以拓展C型肝炎藥物組合。
(5) Jublia® (efinaconazole)
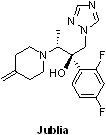 2014年6月6日位於加拿大魁北克的Valeant Pharmaceuticals之灰指甲(onychomycosis)治療藥物Jublia® (efinaconazole, KP-103, 10%外用溶液)獲得FDA核准上市。Jublia®是首個外用型三唑類(triazole)抗真菌藥,屬於14α-demethylase抑制劑,用於紅色毛癬菌(Trichophyton rubrum)和須癬毛癬菌(Trichophyton mentagrophytes)感染的灰指甲治療。灰指甲是一種發生在人指(趾)甲上的傳染性疾病的俗稱,是由病原真菌感染引起,在美國約有35萬名患者。目前非處方或處方外用藥或口服藥物僅提供相當有限的療效,並且用藥的同時往往仍需要頻繁清創,或刮、切割或去除指甲,故整個疾病之有效治療有其侷限性。Jublia®是一種外用溶液,透過一種獨特的內置用量控制型指甲刷進行塗敷,能很快乾燥且無需使用過量藥物,故較無顧慮系統性副作用(如藥物相互作用或急性肝損傷)。
2014年6月6日位於加拿大魁北克的Valeant Pharmaceuticals之灰指甲(onychomycosis)治療藥物Jublia® (efinaconazole, KP-103, 10%外用溶液)獲得FDA核准上市。Jublia®是首個外用型三唑類(triazole)抗真菌藥,屬於14α-demethylase抑制劑,用於紅色毛癬菌(Trichophyton rubrum)和須癬毛癬菌(Trichophyton mentagrophytes)感染的灰指甲治療。灰指甲是一種發生在人指(趾)甲上的傳染性疾病的俗稱,是由病原真菌感染引起,在美國約有35萬名患者。目前非處方或處方外用藥或口服藥物僅提供相當有限的療效,並且用藥的同時往往仍需要頻繁清創,或刮、切割或去除指甲,故整個疾病之有效治療有其侷限性。Jublia®是一種外用溶液,透過一種獨特的內置用量控制型指甲刷進行塗敷,能很快乾燥且無需使用過量藥物,故較無顧慮系統性副作用(如藥物相互作用或急性肝損傷)。Efinaconazole最先是由日本Kaken Pharmaceuticals公司開發,在2006年5月10日技轉給專門開發局部塗抹藥膏的Dow Pharmaceutical Sciences(DPS)公司,在美國和歐洲進行後續臨床開發。然而,在2008年12月10日Valeant Pharmaceuticals以2.85億美元併購DPS,進而取得治療青春痘的上市藥品Acanya®以及承接efinaconazole之開發擁有權,並終於在2014年6月6日取得FDA核准上市。值得一提的是,2013年10月28日,Valeant Pharmaceuticals透過庭外和解,同意支付其競爭同業Anacor Pharmaceuticals公司1.425億美元之損失賠償(原要求2.15億美元賠償金);其中1億美元用於DPS,起因於DPS在2004年承接Anacor公司治療灰指甲藥物tavaborole之製劑委託服務,後於2006年DPS又自行取得日本Kaken治療灰指甲藥efinaconazole之開發權,導致Anacor在智慧財產權、營業秘密以及商業競爭之損失。而另外4,250萬美金為和解金,以解決與2012年12月被Valeant併購的Medicis Pharmaceutical公司之間的爭執。
(6) KerydinTM (tavaborole)
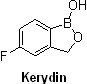 上面提到Anacor公司與Valeant公司之法律爭議,其主角就是KerydinTM(tavaborole,AN2690,5%外用溶液)。KerydinTM於2014年7月7日獲得FDA核准,也是用於紅色毛癬菌和須癬毛癬菌感染的灰指甲治療。FDA的核准是基於2項共納入1,194例患者之臨床試驗結果,每日1次將KerydinTM施用於感染的趾甲,療程共48週,其完全治癒的比例分别為6.5%和9.1%。根據Anacor建議,患者不需要進行趾甲清創術。KerydinTM是第一個oxaborole抗真菌藥,抑制真菌在進行蛋白質合成所需重要酵素(Leucyl-tRNA synthetase, LeuRS),這也是FDA繼通過Jublia®上市,僅僅1個月之後,通過另一個治療灰指甲之外用藥品。但由於KerydinTM的臨床III期結果顯示比Jublia®療效更低(Jublia®完全治癒的比例分别17.8%),故FDA宣布核准隔日,Anacor公司早盤股價大跌近11%,收盤亦跌7%。但Wedbush分析師預測,2015年KerydinTM在美國銷售額可達1,600萬美元,到2021年將達到3.47億美元之銷售高峰。
上面提到Anacor公司與Valeant公司之法律爭議,其主角就是KerydinTM(tavaborole,AN2690,5%外用溶液)。KerydinTM於2014年7月7日獲得FDA核准,也是用於紅色毛癬菌和須癬毛癬菌感染的灰指甲治療。FDA的核准是基於2項共納入1,194例患者之臨床試驗結果,每日1次將KerydinTM施用於感染的趾甲,療程共48週,其完全治癒的比例分别為6.5%和9.1%。根據Anacor建議,患者不需要進行趾甲清創術。KerydinTM是第一個oxaborole抗真菌藥,抑制真菌在進行蛋白質合成所需重要酵素(Leucyl-tRNA synthetase, LeuRS),這也是FDA繼通過Jublia®上市,僅僅1個月之後,通過另一個治療灰指甲之外用藥品。但由於KerydinTM的臨床III期結果顯示比Jublia®療效更低(Jublia®完全治癒的比例分别17.8%),故FDA宣布核准隔日,Anacor公司早盤股價大跌近11%,收盤亦跌7%。但Wedbush分析師預測,2015年KerydinTM在美國銷售額可達1,600萬美元,到2021年將達到3.47億美元之銷售高峰。(7) XtoroTM (finafloxacin)
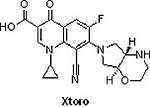 美國FDA於2014年12月17日,依優先審查制度核准愛爾康實驗室(Alcon於2010年已被Novartis併購)的XtoroTM(finafloxacin,otic suspension,耳科滴液)用於治療由綠膿桿菌和金黃色葡萄球菌引起的急性外耳道炎,俗稱「游泳性耳炎(swimmer’s ear)」。急性外耳道炎是發生在外耳和耳道等部位的感染,通常由耳道內的細菌引起,導致疼痛、腫脹、耳朵發紅及耳朵流膿等。耳朵處於水下(如游泳)會使耳道潮濕,細菌容易滋生而發生感染。XtoroTM屬氟喹諾酮類抗菌類藥物,是一種能在酸性條件下被活化,根除幽門螺桿菌的獨特抗生素,其作用機制為DNA gyrase和topoisomerase IV抑制劑。
美國FDA於2014年12月17日,依優先審查制度核准愛爾康實驗室(Alcon於2010年已被Novartis併購)的XtoroTM(finafloxacin,otic suspension,耳科滴液)用於治療由綠膿桿菌和金黃色葡萄球菌引起的急性外耳道炎,俗稱「游泳性耳炎(swimmer’s ear)」。急性外耳道炎是發生在外耳和耳道等部位的感染,通常由耳道內的細菌引起,導致疼痛、腫脹、耳朵發紅及耳朵流膿等。耳朵處於水下(如游泳)會使耳道潮濕,細菌容易滋生而發生感染。XtoroTM屬氟喹諾酮類抗菌類藥物,是一種能在酸性條件下被活化,根除幽門螺桿菌的獨特抗生素,其作用機制為DNA gyrase和topoisomerase IV抑制劑。XtoroTM的安全性和有效性在6個月至85歲之間的1,234例患者參與者的2項臨床試驗獲得了初步確立,參與者被隨機分配接受XtoroTM或賦形劑(用以增加藥物的均勻性、穩定性與減少藥物的刺激性、不良氣味等,而添加在有效成分外的物質)。在其中一個包括560例確認是由綠膿桿菌或金黃色葡萄球菌引起的急性外耳道炎參與者之試驗中,XtoroTM臨床治愈率70%,而賦形劑為37%。
XtoroTM亦如其他大部分感染症藥物,並非由申請上市核准公司Alcon內部開發藥物,而是Alcon於2011年1月11日從2002年成立的新加坡魚尾獅藥劑公司(MerLion Pharmaceuticals)技轉取得治療耳部感染之北美所有權,而其他適應症與專利地區仍屬於MerLion Pharmaceuticals。MerLion Pharmaceuticals為新加坡政府設立的分子與生物細胞研究院(Institute of Molecular and Cell Biology, IMCB)之天然產品研究中心(Centre for Natural Product Research)私有化(privatization)後成立的,是世界主要的天然藥物開發公司,擁有世界最大及最多元化的天然產品樣本收藏,與許多著名藥劑廠和研發機構建立夥伴關係,隨著XtoroTM在美國上市,新加坡亦擠身與日本和南韓一樣,有自行研發產品能夠被美國FDA核准上市。
(8) RapivabTM (peramivir)
 RapivabTM為神經胺酸酶(neuraminidase)抑制劑,可用以治療A型或B型流感病毒感染。2009年10月,已完成臨床I期和II期試驗的RapivabTM正進行臨床III期試驗。與此同時,一種帶有和豬流感(swine flu)基因高度相關的A型流感病毒H1N1亞型,造成人傳人的流行性感冒大爆發(俗稱「豬流感」),這種新流感會導致部分患者出現嚴重併發症甚至死亡,就算在確診後24小時內使用了克流感(Tamiflu®)或瑞樂沙(Relenza®)治療,效果亦不佳。然而,RapivabTM注射劑在當時被報導可以克服抗藥性以及因應嚴重病情,例如昏迷的病人因無法吞服克流感或吸入瑞樂沙之治療需求;研究結果顯示,此新藥可將流感發病期程縮短32%,減輕流感症狀的嚴重程度,減低病毒人傳人的機會,故2009年10月23日FDA給予RapivabTM緊急使用授權(emergency use authorization, EUA),以作為流感大爆發時之最後防線。之後,BioCryst Pharmaceuticals公司在接受美國衛生和公眾服務部(Department of Health and Human Services, HHS)0.77億美元支助下始完成RapivabTM臨床III期試驗。該藥物在2010年1月由Shionogi公司在日本上市,商品名為Rapiacta®;2011年,該藥物在韓國核准,商品名為Peramiflu®,用於治療流感、豬流感和禽流感。
RapivabTM為神經胺酸酶(neuraminidase)抑制劑,可用以治療A型或B型流感病毒感染。2009年10月,已完成臨床I期和II期試驗的RapivabTM正進行臨床III期試驗。與此同時,一種帶有和豬流感(swine flu)基因高度相關的A型流感病毒H1N1亞型,造成人傳人的流行性感冒大爆發(俗稱「豬流感」),這種新流感會導致部分患者出現嚴重併發症甚至死亡,就算在確診後24小時內使用了克流感(Tamiflu®)或瑞樂沙(Relenza®)治療,效果亦不佳。然而,RapivabTM注射劑在當時被報導可以克服抗藥性以及因應嚴重病情,例如昏迷的病人因無法吞服克流感或吸入瑞樂沙之治療需求;研究結果顯示,此新藥可將流感發病期程縮短32%,減輕流感症狀的嚴重程度,減低病毒人傳人的機會,故2009年10月23日FDA給予RapivabTM緊急使用授權(emergency use authorization, EUA),以作為流感大爆發時之最後防線。之後,BioCryst Pharmaceuticals公司在接受美國衛生和公眾服務部(Department of Health and Human Services, HHS)0.77億美元支助下始完成RapivabTM臨床III期試驗。該藥物在2010年1月由Shionogi公司在日本上市,商品名為Rapiacta®;2011年,該藥物在韓國核准,商品名為Peramiflu®,用於治療流感、豬流感和禽流感。2012年BioCryst製藥公司由於RapivabTM臨床III期期中分析結果不佳,一度要終止RapivabTM臨床III期試驗,但在2013年美國生物醫學高級研究開發局(Biomedical Advanced Research and Development Authority, BARDA)以及美國HHS聯合補助2.348億美元下,始順利完成RapivabTM注射劑之臨床III期試驗,並於2013年12月提出NDA,於2014年12月19 日獲得美國FDA核准用於治療成人患者流感。RapivabTM的療效是建立於297例確診流感參與者的試驗,受試者被隨機分配接受RapivabTM 300毫克、RapivabTM 600毫克或安慰劑。總體而言,接受RapivabTM 600毫克治療的參與者比接受安慰劑者平均提前21小時得到流感症狀緩解,且平均提前12小時恢復到正常體溫。
RapivabTM是被FDA核准用於治療流感感染的第3個神經胺酸酶抑制劑,其不僅為第一個批准為IV給藥、單劑量使用的藥物,也是15年來FDA核准的一種全新的流感抗病毒治療方法。據美國CDC的統計,每年有5至20%的美國人會得流感,超過20萬人會因季節性流感併發症而住院,因此Roth Capital Partner的分析師EdArce估計,儘管RapivabTM的商業潛力有限,然而因主要是用於住院患者,通常庫存訂單都會驅動銷量,因此銷售應該不成問題,故2015年第1季,BioCryst會從美國HHS獲得1.2億美元的庫存訂單。
(9) Impavido® (miltefosine)
 2014年3月19日FDA核准位於加拿大蒙特婁Knight Therapeutics公司的Impavido®(miltefosine, Miltex, hexadecylphosphocholine, D-18506)用於治療12歲以上患者的內臟利什曼原蟲症(visceral leishmaniasis)、皮膚利什曼原蟲症(cutaneous leishmaniasis)和黏膜利什曼原蟲症(mucocutaneous leishmaniasis)。Impavido®符合2007年FDA頒布的《熱帶病優先審查憑證(Tropical Disease Priority Review Voucher)》修正法案,是以快速審查資格、優先審評和孤兒藥資格而核准的首項表皮和黏膜利什曼原蟲症藥物。利什曼原蟲症是由利什曼原蟲感染引起,經白蛉叮咬傳播的一種熱帶和亞熱帶寄生蟲傳染人畜共患病,以皮膚或內臟器官的嚴重損害、壞死為特徵。世界衛生組織估計,全世界約有3億5,000萬人感染利什曼原蟲症,每年全世界新增150萬個皮膚型及50萬個內臟型病例。全世界有82個國家被視為流行地區,其中21個國家在新大陸,61個國家在舊大陸。
2014年3月19日FDA核准位於加拿大蒙特婁Knight Therapeutics公司的Impavido®(miltefosine, Miltex, hexadecylphosphocholine, D-18506)用於治療12歲以上患者的內臟利什曼原蟲症(visceral leishmaniasis)、皮膚利什曼原蟲症(cutaneous leishmaniasis)和黏膜利什曼原蟲症(mucocutaneous leishmaniasis)。Impavido®符合2007年FDA頒布的《熱帶病優先審查憑證(Tropical Disease Priority Review Voucher)》修正法案,是以快速審查資格、優先審評和孤兒藥資格而核准的首項表皮和黏膜利什曼原蟲症藥物。利什曼原蟲症是由利什曼原蟲感染引起,經白蛉叮咬傳播的一種熱帶和亞熱帶寄生蟲傳染人畜共患病,以皮膚或內臟器官的嚴重損害、壞死為特徵。世界衛生組織估計,全世界約有3億5,000萬人感染利什曼原蟲症,每年全世界新增150萬個皮膚型及50萬個內臟型病例。全世界有82個國家被視為流行地區,其中21個國家在新大陸,61個國家在舊大陸。過去傳統的藥物治療效果不佳,主要是因為藥物的副作用、效果差、同時有多種疾病及antimony抗藥性株的產生。4項臨床研究評估了患者接受Impavido®(n = 547)、對照藥或安慰劑(n = 183)的療效和安全性,結果顯示口服藥物Impavido®治療內臟、皮膚和黏膜利什曼原蟲症時安全有效。
Miltefosine為蛋白激酶C(PKC)抑制劑,結構類似於細胞膜的磷脂成分,在1980年初期被發現,到1980年後期分別在德國Max Planck Institute for Biophysical Chemistry與University of Göttingen任職的Hansjörg Eibl和Clemens Unger發現miltefosine可以治療乳腺癌,之後在1993年由Baxter Oncology公司將miltefosine針對癌症開發上市。此外,1980年後期任職於London School of Hygiene and Tropical Medicine 的 S. L. Croft博士發現miltefosine可以抗利什曼原蟲,且因之後在印度進行臨床試驗安全性不錯,進而啟動ASTA Medica公司(後來為Zentaris GmbH)、WHO Special Programme for Research and Training in Tropical Diseases,以及印度政府三方合作,在2002年完成臨床II期和III期試驗,2003年由Zentaris GmbH負責在印度、哥倫比亞和德國推動上市。爾後,德國Zentaris GmbH變成加拿大魁北克AEterna Zentaris公司,以開發癌症和內分泌疾病藥物為專長,故在2008年3月3日以920萬美金將Impavido®後續權利賣給位於蒙特婁的Paladin實驗室。接者,Paladin實驗室於2013年11月5日以16億美金被位於愛爾蘭都柏林的Endo Health Solutions公司(原為美國公司)併購,但當時Paladin實驗室CEO Jonathan Goodman透過與Endo Health Solutions磋商,取得Impavido®後續開發權,而成立新創公司Knight Therapeutics,最後由Knight Therapeutics於2014年3月19日順利將Impavido®推上美國市場。
註1:新藥大放異彩-漫談2014年FDA核准小分子新藥的報導,主要是根據註3至註5這3篇文章所提供之資訊撰寫。
註2:二十多年前,一款新藥若要通過FDA的審查,從遞交申請到核准上市平均需花費30個月,相當冗長耗時。然,美國國會於1992年通過《處方藥使用者費用法案(Prescription Drug User Fee Act, PDUFA)》,送審業者必須支付審查費用給FDA,這筆user fee可以讓FDA聘僱更多專業人士,不僅加快審核的速度,也提升專業水準。《處方藥使用者費用法案》最近一次的更新在2012年,目前新藥從審核到上市平均只需15個月,而治療某些疾病的藥物如愛滋病,更能優先審核,平均只需6個月。
註3:Morrison, C. Fresh from the biotech pipeline-2014. Nature Biotechnology. 2015, 33, 125-128.
註4:Jarvis, L. M. The year in new drugs. Chemical & Engineering News. 2015 February 2, 11-16.
註5:Mullard, A. 2014 FDA drug approvals. Nature Reviews Drug Discovery. 2015, 14, 77-81.
新藥大放異彩-漫談2014年FDA核准小分子新藥(上)。《國家衛生研究院電子報607期》。
《文/圖:生技與藥物研究所謝興邦研究員、李靜琪副研究員;審校:生技與藥物研究所陳炯東研究員》