NHRI Communications

研究發展
發炎因子促進口腔細胞增生與癌化:促發炎細胞激素IL-1β轉活化EGFR而促進癌前細胞增生
NHRI researcher reports interleukin-1 beta transactivates epidermal growth factor receptor via the CXCL1-CXCR2 axis in oral cancer
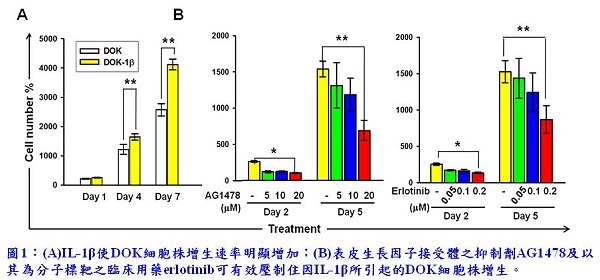
表皮生長因子接受體(epidermal growth factor receptor, EGFR)過度表現或活化常見於口腔鱗狀細胞癌,有文獻指出高於八成的口腔癌組織皆有EGFR過度表現或過度活化的現象,且此異常現象在口腔癌變前期的細胞中即已發生。EGFR的異常活化會使其下游的細胞訊息傳導途徑失控,因而促使細胞不正常增生,甚至癌化。先前本院癌症研究所李家惠博士實驗室的研究發現,口腔鱗狀細胞癌的細胞株皆會分泌高濃度的介白素IL-1β,而香菸致癌物NNK也會刺激口腔細胞分泌IL-1β;IL-1β會大幅增加口腔癌變前期細胞株(dysplastic oral keratinocyte, DOK)的生長速率(圖1(A)),並且加速口腔癌細胞轉移能力。此外,IL-1β亦誘導DOK及口腔癌細胞株分泌趨化因子CXCL1。CXCL1的細胞接受器為CXCR2,為G蛋白偶和接受器(G protein-coupled receptor),在卵巢癌及肺癌細胞中曾被發現會轉活化(transactivates)EGFR;因此,李家惠博士提出假說,認為IL-1β有可能經由誘導口腔細胞分泌CXCL1,而透過CXCL1活化CXCR2再由CXCR2轉活化EGFR,終致口腔癌前病變細胞增生且促使其癌化。為了評估這假說的可能性,李博士實驗室先以EGFR之抑制劑AG1478及以之為分子標靶的臨床用藥erlotinib測試,實驗結果顯示AG1478和erlotinib對於IL-1β所引起的口腔癌前病變細胞株DOK的增生有顯著的抑制效果(圖1(B)),這意味著EGFR的活化可能與IL-1β所促使的細胞增生有關。
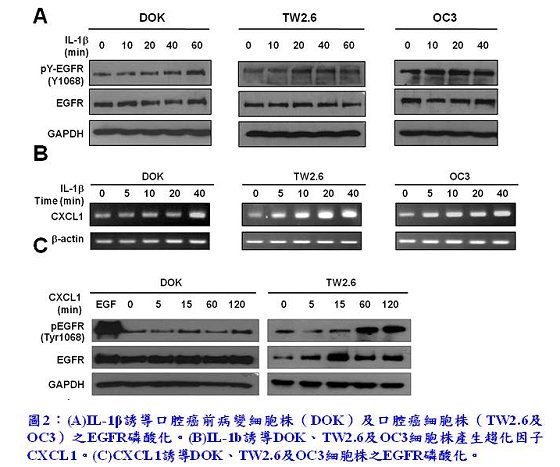
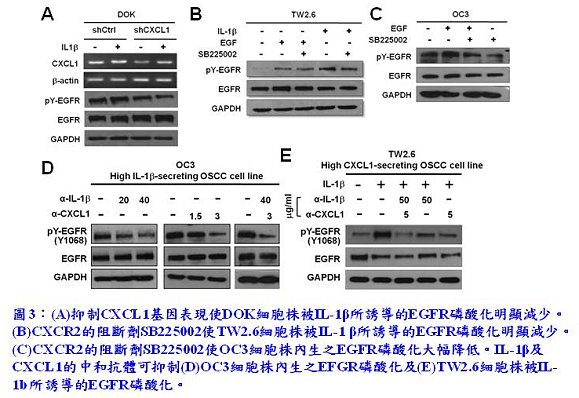
接續的一連串實驗證實,IL-1β在口腔癌及癌變前期細胞株中皆能誘導EGFR磷酸化及產生CXCL1(圖2(A)及(B)),而外加的CXCL1亦可誘導EGFR磷酸化(圖2(C))。同時,也發現分泌高量IL-1β的本土口腔癌細胞株OC3其EGFR之內生性(intrinsic)磷酸化遠比分泌相對低量IL-1β的本土口腔癌細胞株TW2.6及DOK來得強,暗示臨床上有可能以唾液中IL-1β的濃度作為口腔癌的生物標誌(biomarker),例如用此非侵入性的檢測,篩檢出癌細胞對EGFR訊息傳遞倚賴的口腔癌患者。接著,為了更加確定IL-1β、CXCL1-CXCR2及EGFR活化這三者間的關係,李博士實驗室進一步利用以下3種方法求證IL-1β與CXCL1-CXCR2軸對EGFR磷酸化的影響:(1)慢病毒感染法專一性抑制CXCL1的基因表達(圖3(A));(2)CXCR2的阻斷劑SB225002(圖3(B));(3)IL-1β或CXCL1的中和抗體(圖3(C))。實驗結果顯示無論是在DOK細胞或TW2.6細胞中,由IL-1β所誘導的EGFR磷酸化,或是在OC3細胞中內生性的EGFR磷酸化,皆可經由以上3種方式的處理而被有效抑制。這結果似乎也暗示著對於EGFR依賴的口腔癌細胞而言,IL-1β、CXCL1和CXCR2具有作為腫瘤標靶治療標靶分子的潛力。
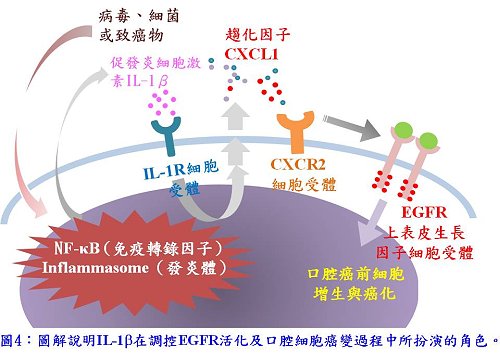 最後,李博士依據這些實驗結果提出「發炎促進口腔細胞癌化進程」之模式(如圖4所示):首先是口腔細胞受某些特定的病原體感染或暴露於致癌物(如NNK)中,因而引發口腔細胞分泌IL-1β,IL-1β透過其接受體IL-1R產生自泌效應(autocrine)使口腔細胞分泌CXCL1,CXCL1與其受體CXCR2結合後,CXCR2進而轉活化了EGFR,因而啟動了EGFR下游的訊息傳導途徑,這些訊息傳導途徑的失調促使了口腔癌前病變細胞的增生,也可能導致口腔癌細胞的惡化。在這個途徑中,CXCL1-CXCR2軸扮演傳輸者的關鍵角色,透過CXCL1-CXCR2軸將許多會誘導細胞產生促發炎的IL-1β分泌之病原體(如病毒或細菌之基因體或蛋白)或環境中存在的化合物(如NNK)轉化為致癌性的細胞訊息傳遞網,此發現不僅再次強調了發炎與癌症的關聯,也為許多與癌症相關的病原體及化合物的致癌機轉提出另一種可能性。
最後,李博士依據這些實驗結果提出「發炎促進口腔細胞癌化進程」之模式(如圖4所示):首先是口腔細胞受某些特定的病原體感染或暴露於致癌物(如NNK)中,因而引發口腔細胞分泌IL-1β,IL-1β透過其接受體IL-1R產生自泌效應(autocrine)使口腔細胞分泌CXCL1,CXCL1與其受體CXCR2結合後,CXCR2進而轉活化了EGFR,因而啟動了EGFR下游的訊息傳導途徑,這些訊息傳導途徑的失調促使了口腔癌前病變細胞的增生,也可能導致口腔癌細胞的惡化。在這個途徑中,CXCL1-CXCR2軸扮演傳輸者的關鍵角色,透過CXCL1-CXCR2軸將許多會誘導細胞產生促發炎的IL-1β分泌之病原體(如病毒或細菌之基因體或蛋白)或環境中存在的化合物(如NNK)轉化為致癌性的細胞訊息傳遞網,此發現不僅再次強調了發炎與癌症的關聯,也為許多與癌症相關的病原體及化合物的致癌機轉提出另一種可能性。以上研究成果榮獲今年台灣癌症聯合學術年會、美洲華人生物科學學會第15屆國際學術研討會的優等論文壁報獎,也獲得院內舉辦的2015年研發成果招商海報競賽優等獎;相關論文也於近日為國際期刊Oncotarget所接受。
《文/圖:癌症研究所李家惠助研究員》