NHRI Communications

知識產權
美國食品藥物管理局(FDA)與歐洲藥物管理局(EMA)新藥審查制度之差異比較初探
Comparisons of U.S. Food and Drug Administration and European Medicine Agency new drug review system
 今(2016)年初,法國爆發歷來最嚴重的藥物試驗事故,導致1人腦死、5人生命垂危,其中有3人腦部受到永久損害,此試驗新藥是為了治療情緒紊亂、焦慮症以及神經退化疾病(阿滋海默症)等[1]。
今(2016)年初,法國爆發歷來最嚴重的藥物試驗事故,導致1人腦死、5人生命垂危,其中有3人腦部受到永久損害,此試驗新藥是為了治療情緒紊亂、焦慮症以及神經退化疾病(阿滋海默症)等[1]。調查發現,臨床前試驗中,曾有試驗動物因腦部傷害而死亡,然而該公司基於機密等考量而未公開 [2]。雖然初步報告認為作業流程上應無疏失,但其他獨立報告仍發現於臨床試驗流程與溝通上的疏失,導致此不可逆傷害的擴大發生[3]。因此,若能更嚴謹地設計、執行與監督臨床前安全性試驗與臨床人體試驗之流程,應可避免傷害的發生。
由於美國食品藥物管理局(US Food and Drug Administration, FDA)與歐洲藥物管理局(European Medicine Agency, EMA)在法規制度上的差異,可能造成藥害風險的不同。在新藥審查制度的案例中,沙利竇邁(thalidomide)悲劇是最為人所知的藥害案例,也開啟了現代新藥審查制度的思維與機制。1960年代,歐洲及日本等多國核准之新藥沙利竇邁,主要用於舒緩孕婦妊娠反應所導致的孕吐症狀,事後追蹤發現藥物造成上千位罕見的海豹肢畸形(phocomelia)新生兒,隨即於1961年被禁用[4];歸功於FDA嚴謹之新藥申請與核准流程,該藥在美國當時尚未獲准用於孕婦[5],此案例說明新藥進入市場前對於其安全性與有效性的評估同樣重要。該事件發生後,促使1960年代至1970年代各國迅速立法規範,1962年FDA率先訂定實證(evidence-based)的藥品評估機制,規定藥廠必須提出證據並說明該藥品同時具備安全性與有效性;並進一步地頒布《優良實驗室操作規範(Good Laboratory Practice, GLP)》以確保臨床前藥物安全性與毒理試驗原始數據之可靠性與完整性,進而促使現今申請新藥臨床試驗至上市之監管機制與系統的建立[6]。
市場的國際化是製藥產業發展的趨勢及擴大獲利的必要條件。雖然嚴謹的法規可以保障民眾用藥安全,基於各國藥物監管機關對於技術資料的需求不同,產業界須投入更多時間、經費與人力以符合各國不同法規的需求,對於急需新藥治療之疾病因藥物上市的延宕,導致損害患者之治療權益。因審查延誤可能造成醫療照護成本的相對增加,以癌症照護花費為例,癌症照護之醫藥花費逐年增長,根據統計,2009年歐洲國家統計約為510億歐元[7];而2010年在美國癌症照護醫藥費用約為1,200億美元[8],高昂的新藥價格也造成患者的治療負擔,整體的醫療照護成本也提升。因此,全球迫切需要統合新藥審查監管環境,使新藥審查的時程精簡而確實,採多元化審核途徑與方式,使更多新藥可以盡快進入市場,但仍需要有效規範與廣泛應用的監管機制。有鑑於此,「國際醫藥法規協和會(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, ICH)」組織成立,其前身為1990年成立之International Conference on Harmonisation(ICH),FDA及EMA為其主要成員[9],從基本架構上來看,ICH的設計十分接近FDA的架構[10]。經過二十餘年努力,兩方的新藥審查監管機構均有很大進步,諸如FDA與EMA雙方分享資訊、國際合作稽查(International API Inspection Pilot Program)或新ICH組織的成立等;然而,FDA與EMA仍有差異,導致研究資源的消耗,主要來自於藥物核准所需的時間期程、用藥指示用詞差異、審查流程中對於諸如患者報告效果(patient-reported outcomes, PROs)審核評估看法不同,以及審查類型比例不同等。以下將依上述幾個方向分析FDA與EMA在新藥審查機制上之差異。
新藥審查時程之差異
每一個成功的新藥,從研究發展至上市平均約需投入18億美元[11],在此之前,投入的資源尚無法開始獲利,患者亦無法獲得新的治療選擇,因此,能越快進入市場,新藥才能得到實質效益。2015年,湯森路透集團之國際法規科學創新研究中心(Centre for Innovation in Regulatory Science, CIRS)所發表之《R&D Briefing 57》報告中,分析了2005至2014年之間EMA與FDA之新藥批准趨勢。報告顯示,2014年審核時間中位數分別為418天(EMA)及343天(FDA), FDA之變化較EMA小,近三年之審核所需時間較為穩定[12]。然而,EMA與FDA所需時間差異主要來自於兩者之主管機關的審核權限不同,FDA同時負責新藥之科學評估及市場銷售之授權許可;而EMA之科學專家委員會之人用藥物委員會(Committee for Medicinal Products for Human Use, CHMP)之權限僅於科學評估。依據CHMP之評估結果,再由EMA提出建議經歐洲議會(European Commission, EC)核准後進入市場銷售。由於歐洲的科學評估與上市銷售授權的審核機制不連貫,造成審查與核准上市的時間延誤。根據統計,12個酪氨酸激酶抑制劑(tyrosine kinase inhibitors, TKIs)藥物中,EMA所需時間約為FDA之兩倍(410天vs. 205天),原因來自於:(1)審評暫停(clock stop),以便澄清審查過程中提出之疑議;(2)自CHMP評估完成後,到最終得到歐洲議會核准前所花費的時間,平均約90天的延遲[13,14]。
比較EMA與FDA對於一個癌症新藥審查平均所需的時間(如圖1所示),EMA審查一個新藥所需時間比FDA平均多花費6個月,主要在於取得CHMP正面意見與等待歐洲議會核准等時間。此外,幾乎全部癌症新藥均可在美國透過FDA的優先審查(priority review)機制被核准,而EMA卻鮮少運用類似此快速審查的機制[14]。
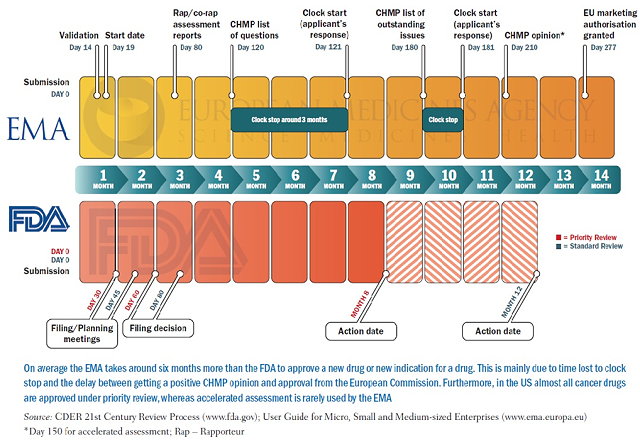
圖1:FDA與EMA新藥核准時程比較(圖片來源:參考文獻 [15])。
用藥指示用詞(wording of indications)之差異
定義治療適應症範圍是藥品監管的一個關鍵步驟,用藥指示用詞可能決定納入或排除某些患者族群,因此對患者影響甚鉅。根據Journal of Clinical Oncology於2011年的一份研究[15],研究人員收集了1995至2008年間EMA核准的42種抗癌新藥所對應之100條用藥指示,其中,53條用藥指示與FDA核准的相同,19條用藥指示不被FDA所核可,有28條用藥指示與FDA核准的不同。值得注意的是,FDA與EMA中之一方首次核准後,另一方往往會較限縮用藥指示用詞或治療適應症範圍,此現象是造成雙方差異之主因。降低FDA與EMA雙方對於指示用詞核准標準差異,以增進患者權益,是有待努力的方向。
患者報告結果(patient-reported outcomes, PROs)的評估重點不同
患者報告結果係直接由患者端經由合理的問卷量表記錄所取得之疾病與治療狀況相關的評估結果,包含患者對於疾病症狀或治療之不良作用、健康相關生活品質(health-related quality of life, HRQL)、健康狀態、藥物治療依從性(adherence to treatment)以及治療滿意度等,此為患者自身評估測量結果,不經他人修改或解釋[16]。EMA與FDA雙方均肯定PROs在評估藥效及決定藥物是否核准之重要性,但雙方採取不同處理方式。EMA著重於HRQL多面向評估測量,而FDA則較重視症狀為主(symptom-specific)的測量,且PROs於FDA常被視為臨床藥效的補充證明之一,因此,相對於EMA,FDA之PROs相關說明文件要求更為嚴格[17]。由於FDA與EMA對於PROs的評估重點不同,因此被FDA或EMA任一方所採用之PROs可能會遭另一方拒絕,這是規劃申請新藥開發時,若欲向FDA與EMA雙方均提出申請,須納入考量的部分。
審查類型策略運用不同
CIRS於2015年之《R&D Briefing 57》報告指出EMA審核批准新藥之期程略長於FDA所需時間[12],除了因雙方權責機關制度不同外,其審查類型亦有很大的影響。加速審查監管路徑(facilitated regulatory pathways, FRPs)為加速審查或批准藥品上市的監管路徑,目的為加快學名藥審查並加速罕病孤兒藥、重症疾病所需藥物或尚無有效治療方法之疾病的藥物需求等,提供別於標準審查的替代審查途徑。《R&D Briefing 57》報告中進一步分析:FDA與EMA於2005年至2014年間,經由標準審查(standard review)與快速審查(expedited review;EMA為加速核准(accelerated assessment);FDA為優先審查(priority review))不同的審查監管路徑所取得的新藥核准數占比不同,FDA(58%)較EMA(僅13%)通過的案例為高[12]。相對於FDA,EMA對於加速審查監管路徑(FRPs)的標準更為嚴格且複雜,因此不常被使用。
EMA於2016年3月7日公告新藥審查的快速通道—優先藥物(priority medicines, PRIME [18])與FDA於2012年7月9日所實施之「突破性治療藥物認證(breakthrough therapy designation, BTD)」[19,20] 具有相近的立法精神,宗旨在於使患者能及早使用到創新藥物,從研發到上市的過程中,全程獲得專業的輔導與主管機關的支持,經由選定擁有臨床前或臨床試驗的初步科學證據,且提供證明為滿足現行醫療不足之迫切需求以及公共健康利益的藥物,主管機關得早期介入協助該藥物開發,提供臨床前或臨床試驗設計之建議與法規諮詢,較有效率地協助藥廠進行藥物發展,加速滿足治療藥物的迫切需求。近期FDA與EMA分別透過突破性治療藥物認可與優先藥物之法規平台展開合作,雙方基於保密協定互換彼此已通知審查之新藥物質清單,因此,預期被優先藥物所核准的藥物,亦有較高機會被突破性治療藥物認可所接受,反之亦然[21]。雖然具體細節尚未清楚,但已為往後的合作概念與平台奠立好的開始。
EMA與FDA之法規環境條件不同,其間之新藥審查程序也有差異。綜合上述,FDA審查與EMA相異之處包括:平均所需期程較短、快速審查管道運用更靈活、對於藉由科學證據說明藥物安全性及療效較為重視。然而,因全球化趨勢所需,雙方已正視因為法規差異所導致新藥研發成本的上升,而影響患者與政府於健康照護費用與負擔加重的問題,並加速努力尋求整合。若藥品需進軍國際市場,則必須以ICH的認證規範為標準[10]。基於ICH的精神,EMA與FDA亦會隨時依未來需要而適度修正審查規範,為各國藥廠以及藥物監管機關之主要參考依據。相信在新藥研發過程符合FDA或ICH的規範,有助於縮短藥廠在主要市場之臨床試驗與審查時程,亦對藥品進軍國際市場有直接的助益,最終將能提高人類的健康福祉。
參考文獻
- 閻紀宇。1人腦死、5人命危 歐洲新藥人體臨床試驗出現恐怖差錯。風傳媒。2016。
- Willsher, K. French drug trial scandal: “dogs died in pre-clinical test”, in the Guardian. 2016, Guardian News and Media Limited.
- Bichell, R.E. Botched French drug trial followed rules but lacked common sense, in National Public Radio (NPR). 2016.
- 李尚仁。沙利竇邁悲劇半世紀。《科學發展月刊》,2015;511: 82-85。
- Scheindlin, S. The courage of one’s convictions: the due diligence of Frances Oldham Kelsey at the FDA. Molecular interventions, 2011. 11(1): 3.
- 徐立峰。藥物研發面面觀:藥物的上市登記與管理。《科學發展月刊》。2015; 505: 6-11.
- Luengo-Fernandez, R., et al. Economic burden of cancer across the European Union: a population-based cost analysis. Lancet Oncol, 2013;14(12): 1165-74.
- Mariotto, A.B., et al. Projections of the cost of cancer care in the United States: 2010-2020. J Natl Cancer Inst, 2011;103(2): 117-28.
- ICH. The Need to Harmonise. [cited 2016 May 5th]; Available from: http://www.ich.org/about/history.html.
- 杜蕙蓉。《生技醫療》認識FDA (四)。2011 [cited 2015 May 5th]; Available from: http://www.taimedbiologics.com.tw/news-18.aspx.
- Reynolds, K.S. Acceleration of drug development: a collaboration of many stakeholders. Clin Pharmacol Ther, 2013;93(6): 455-9.
- Bujar, M., McAuslane, N., and Liberti, L. New drug approvals in ICH countries 2005 – 2014. Focus on facilitated regulatory pathways and orphan designations (R&D Briefing 57). 2015, Centre for Innovation in Regulatory Science (CIRS), London.
- Shah, R.R., S.A. Roberts, and D.R. Shah, A fresh perspective on comparing the FDA and the CHMP/EMA: approval of antineoplastic tyrosine kinase inhibitors. Br J Clin Pharmacol, 2013;76(3): 396-411.
- Beishon, M. Approval rating: how do the EMA and FDA compare? CancerWorld, 2014;12-17.
- Trotta, F., et al., Evaluation of oncology drugs at the European Medicines Agency and US Food and Drug Administration: when differences have an impact on clinical practice. J Clin Oncol, 2011;29(16): 2266-72.
- 董淑敏。歐盟EMA於2014年6月發表「腫瘤學研究使用病人報告效果(Patient Reported Outcome; PRO)測量」觀點討論。《財團法人醫藥品查驗中心—當代醫藥法規》, 2014;45: 26.
- Howie, L.J., B.R. Hirsch, and A.P. Abernethy. A comparison of FDA and EMA drug approval: implications for drug development and cost of care. Oncology (Williston Park), 2013;27(12): 1195, 1198-1200, 1202 passim.
- EMA. Launch of PRIME – Paving the way for promising medicines for patients. 07/03/2016 [cited 2016 May 10th]; Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2016/03/news_detail_002484.jsp&mid=WC0b01ac058004d5c1.
- Ledford, H., Pharma scrambles to fast-track drugs. Nature, 2013;502(7469): 20.
- FDA. Fact Sheet: Breakthrough Therapies. 12/10/2014 [cited 2016 May 10th]; Available from: http://www.fda.gov/RegulatoryInformation/Legislation/SignificantAmendmentstotheFDCAct/FDASIA/ucm329491.htm.
- Sharma, V., EMA, FDA get together on drugs eligible for prime and breakthrough designation. The Pink Sheet, 2016.
《文/圖:生技與藥物研究所徐嘉瑜博士後研究員、陳炯東研究員》