NHRI Communications

知識產權
新藥再創新猷—漫談2015年美國FDA核准小分子新藥(上)
Introduction to FDA approved novel small molecular drugs in 2015 (part 1)
 延續2014年生技醫藥產業於新藥研發的歡喜豐收「註1」,2015年更甚於2014年,共有45個新藥獲得美國食品藥物管理局(U.S. Food and Drug Administration, U.S. FDA)核准上市,包括32個小分子藥物和13個生物藥(抗體藥物、胜肽藥物及酶)「註2」。自2014年的榮景即可見FDA對新藥申請審查效率的提升,而「More important than the quantity of novel drugs approved by Center for Drug Evaluation and Research (CDER) in 2015 is their quality and the important new roles they are serving to advance medical care」說明了在效率提升的同時,藥物的療效和安全性及促進醫療保健的角色依然是CDER在新藥審查時最關心的部分「註3」。
延續2014年生技醫藥產業於新藥研發的歡喜豐收「註1」,2015年更甚於2014年,共有45個新藥獲得美國食品藥物管理局(U.S. Food and Drug Administration, U.S. FDA)核准上市,包括32個小分子藥物和13個生物藥(抗體藥物、胜肽藥物及酶)「註2」。自2014年的榮景即可見FDA對新藥申請審查效率的提升,而「More important than the quantity of novel drugs approved by Center for Drug Evaluation and Research (CDER) in 2015 is their quality and the important new roles they are serving to advance medical care」說明了在效率提升的同時,藥物的療效和安全性及促進醫療保健的角色依然是CDER在新藥審查時最關心的部分「註3」。2015年U.S. FDA所核准的45個新藥,依其特點可歸類如下「註3」:(a) 36%(16個)是具新機轉的市場首見(first-in-class)新藥;(b) 47%(21個)是用於治療患者總數少於20萬人的罕見疾病用藥;(c) 60%(27個)是獲得一種或一種以上加速方案:快速審查(fast track)、突破性藥物(breakthrough)、優先審查(priority review)及加速核准(accelerated approval)資格的新藥;(d) 87%(39個)為首輪核准(first cycle approval)新藥,意即初審就被核准,無須再補充其他資料;(e) 64%(29個)是優先在美國上市的新藥(圖一)。
自疾病領域來看,癌症相關藥物仍然領先群倫,一共有14 個抗癌藥物與1個癌症化療止吐藥獲准上市,占所有新藥的33%,其中包括有4個單株抗體藥物(EmplicitiTM、DarzalexTM和Unituxin® 是first-in-class新藥,另一為Portrazza® );過去4年來,僅2014年的核准比例(22%)未超過30%。除了抗癌藥物延續2014年的風光外,其他領域則互有消長,其中以感染性疾病治療藥物變化幅度最大,由2014年的27%降為2015年9%。本系列報導將分為上中下三期來介紹2015年獲U.S. FDA核准的小分子藥物,於本期率先登場的是癌症相關藥物。
癌症相關藥物
2015年共有14 個抗癌藥物與1個癌症化療止吐藥(Varubi® )獲准上市,其中有4個抗癌藥物是單株抗體藥物(EmplicitiTM 、DarzalexTM 、Portrazza® 、Unituxin® ),以下將針對11個小分子部分詳加介紹「註4 」。
(1) Alecensa® (alectinib)
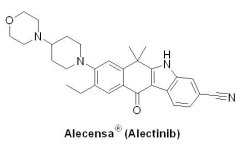 繼2011年9月26日Pfizer藥廠第一個針對間變性淋巴瘤激酶(anaplastic lymphoma kinase, ALK)抑制劑Xalkori® (crizotinib)上市以及2014年4月29日Novartis藥廠開發的第二代ALK抑制劑ZykadiaTM(ceritinib, LDK378)上市後,U.S. FDA於2015年12月11日核准第三個ALK抑制劑,是由Roche藥廠研發的Alecensa® (alectinib, AF802, CH5424802, RG7853, RO5424802),並獲得U.S. FDA授予孤兒藥地位(orphan drug status)、突破性藥物資格和優先審查資格。Alectinib® 用於治療晚期(轉移性)ALK陽性非小細胞肺癌(non-small cell lung cancer, NSCLC)患者,患者曾接受Xalkori® 治療後病情惡化或不能耐受Xalkori® 治療。
繼2011年9月26日Pfizer藥廠第一個針對間變性淋巴瘤激酶(anaplastic lymphoma kinase, ALK)抑制劑Xalkori® (crizotinib)上市以及2014年4月29日Novartis藥廠開發的第二代ALK抑制劑ZykadiaTM(ceritinib, LDK378)上市後,U.S. FDA於2015年12月11日核准第三個ALK抑制劑,是由Roche藥廠研發的Alecensa® (alectinib, AF802, CH5424802, RG7853, RO5424802),並獲得U.S. FDA授予孤兒藥地位(orphan drug status)、突破性藥物資格和優先審查資格。Alectinib® 用於治療晚期(轉移性)ALK陽性非小細胞肺癌(non-small cell lung cancer, NSCLC)患者,患者曾接受Xalkori® 治療後病情惡化或不能耐受Xalkori® 治療。Alecensa® 最早是由日本Chugai Pharmaceutical公司的Kamakura Research Laboratories所開發(Kamakura Research Laboratories原屬於Nippon-Roche Research Center);2001年12月10日Roche以12.3到15.8億美元取得50.1% Chugai Pharmaceutical股票,目前則擁有59.9% Chugai Pharmaceutical股票。Alecensa® 在2014年7月4日,以臨床試驗I/II期(AF-001JP)優異結果獲得日本厚生勞動省新藥核准上市,而在美國部分,FDA以NP28761(87位經Xalkori® 治療復發患者)和NP28673(138位經Xalkori® 治療復發患者)兩個臨床試驗,分別有38%和44%受試者達症狀反應,並且可平均維持7.5個月和11.2個月之結果核准Alecensa® 上市。由於Alecensa® 具較佳的穿透血腦屏障(blood-brain barrier, BBB)能力,有61%腫瘤轉移到腦部的患者在投予Alecensa® 治療後,腫瘤消失或縮小,且平均可維持9.1個月。Alecensa® 初估1個月藥價為1.25萬美元,年銷售額將達10億美元。目前Roche藥廠也正展開一項head-to-head臨床III期試驗,比較Alecensa® 和Xalkori® 分別為一線治療ALK陽性NSCLC的療效和安全性。如果該研究獲得成功,將威脅Pfizer藥廠的Xalkori® 在ALK陽性NSCLC領域的霸主地位。
(2) TagrissoTM (osimertinib)
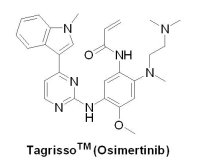 自Iressa® (gefitinib)和Tarceva® (erlotinib)針對上皮細胞生長因子受體(epidermal growth factor receptor, EGFR)基因變異上市後,開啟NSCLC個體化治療之濫觴,爾後在2013年7月由Boehringer Ingelheim公司開發的第二代EGFR抑制劑Gilotrif® (afatinib)獲得U.S. FDA核准上市,由於Gilotrif®對於原生型(wild-type)和突變型(mutation) EGFR不具有選擇性,以致其治療指數(therapeutic index)較低。2015年11月13日,AstraZeneca藥廠開發的第三代EGFR抑制劑TagrissoTM(osimertinib, mereletinib, AZD9291)獲得U.S. FDA核准上市。TagrissoTM是一種口服的、不可逆的第三代EGFR抑制劑(EGFR-TKI),能同時對付NSCLC的EGFR基因突變(包括exon 18、19、21上的突變)和EGFR雙突變(L858R/T790M),並且對於原生型EGFR有較高的選擇性;因此比第一代和第二代EGFR抑制劑擁有較小的副作用。TagrissoTM獲U.S. FDA授予孤兒藥地位、突破性藥物資格、優先審查資格和加速核准資格,比預定期限提前了3個月上市,用於治療EGFR上T790M基因突變或對其它EGFR抑制劑產生抗藥性的晚期NSCLC。
自Iressa® (gefitinib)和Tarceva® (erlotinib)針對上皮細胞生長因子受體(epidermal growth factor receptor, EGFR)基因變異上市後,開啟NSCLC個體化治療之濫觴,爾後在2013年7月由Boehringer Ingelheim公司開發的第二代EGFR抑制劑Gilotrif® (afatinib)獲得U.S. FDA核准上市,由於Gilotrif®對於原生型(wild-type)和突變型(mutation) EGFR不具有選擇性,以致其治療指數(therapeutic index)較低。2015年11月13日,AstraZeneca藥廠開發的第三代EGFR抑制劑TagrissoTM(osimertinib, mereletinib, AZD9291)獲得U.S. FDA核准上市。TagrissoTM是一種口服的、不可逆的第三代EGFR抑制劑(EGFR-TKI),能同時對付NSCLC的EGFR基因突變(包括exon 18、19、21上的突變)和EGFR雙突變(L858R/T790M),並且對於原生型EGFR有較高的選擇性;因此比第一代和第二代EGFR抑制劑擁有較小的副作用。TagrissoTM獲U.S. FDA授予孤兒藥地位、突破性藥物資格、優先審查資格和加速核准資格,比預定期限提前了3個月上市,用於治療EGFR上T790M基因突變或對其它EGFR抑制劑產生抗藥性的晚期NSCLC。TagrissoTM是AstraZeneca藥廠的Richard A. Ward博士在2009年受到哈佛Dana-Farber Cancer Institute的P. A. Jänne教授於《Nature雜誌》所報導之不可逆、並對原生性 EGFR具有高選擇性的化合物WZ-4002之啟發後,領導藥物化學團隊,透過內部篩選及電腦輔助藥物設計而得到。TagrissoTM在2013年開始進行臨床試驗,當時在美國臨床腫瘤醫學會(American Society of Clinical Oncology, ASCO)年度會議,其進度還遠遠落後競爭對手—美國 Clovis Oncology公司的rociletinib(CO-1686),但是在 2014 年 5 月 30 日的 ASCO 年會上,TagrissoTM之進展已經趕上對手的發展,終於在2015年11月率先在第三代EGFR戰場上拔得頭籌。
U.S. FDA核准TagrissoTM是基於2個臨床II期研究(AURA擴展研究及AURA2研究)以及AURA I期擴展研究的積極數據:在474位接受EGFR-TKI治療或治療後病情惡化的EGFRT790M NSCLC患者的臨床結果顯示,TagrissoTM治療組有66%患者的腫瘤完全消除或部分縮小,無惡化存活時間(progression-free survival, PFS)為9.7個月。目前TagrissoTM在臨床使用前必須先檢測患者是否發生T790M基因突變(CobasEGFR突變檢測v2,由美國Roche Diagnostics公司生產),只有T790M基因突變的NSCLC患者才能使用TagrissoTM展開治療。患者每日服用1次TagrissoTM,每次80毫克,1個月藥價為1.275萬美元,1年藥價為15.3萬美元。全球商業數據公司湯森路透預測TagrissoTM在2019年的潛在銷售額為7.61億美元,若是最終能核准成為EGFR突變肺腺癌的一線用藥,其銷售峰值有望達10到30億美元之間。
(3) Cotellic® (cobimetinib)
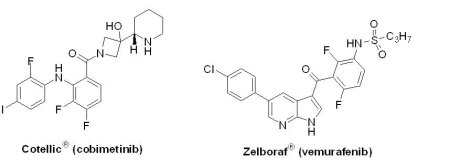 黑色素瘤是一種高度惡性的皮膚腫瘤,易轉移,發病率占所有腫瘤的1至3%,年增長率為3至5%。Mitogen-activated protein kinase kinase(MEK)是一種蛋白激酶靶點,是RAS-RAF-MEK-ERK訊息傳遞路徑的一部分,該通路可促進細胞的分裂和存活,在人類癌症(包括黑色素瘤)中往往處於持續活化狀態。繼2014年1月GlaxoSmithKline(GSK)藥廠開發的Tafinlar® (dabrafenib)合併Mekinist® (trametinib)使用,獲U.S. FDA第一個通過用於治療攜帶BRAF V600E或V600K突變的不可切除性或轉移性黑色素瘤患者。U.S. FDA授予孤兒藥地位和優先審查資格的MEK抑制劑Cotellic® (cobimetinib, XL-518, GDC-0973, RG7420)合併BRAF抑制劑Zelboraf® (vemurafenib,2011核准)緊接著於2015年11月10日獲准上市,是由Roche藥廠所開發的組合藥物。Cotellic® 是Roche藥廠在過去5年中獲得U.S. FDA核准的第7個新藥,此藥物也在15天後獲得歐盟核准上市。
黑色素瘤是一種高度惡性的皮膚腫瘤,易轉移,發病率占所有腫瘤的1至3%,年增長率為3至5%。Mitogen-activated protein kinase kinase(MEK)是一種蛋白激酶靶點,是RAS-RAF-MEK-ERK訊息傳遞路徑的一部分,該通路可促進細胞的分裂和存活,在人類癌症(包括黑色素瘤)中往往處於持續活化狀態。繼2014年1月GlaxoSmithKline(GSK)藥廠開發的Tafinlar® (dabrafenib)合併Mekinist® (trametinib)使用,獲U.S. FDA第一個通過用於治療攜帶BRAF V600E或V600K突變的不可切除性或轉移性黑色素瘤患者。U.S. FDA授予孤兒藥地位和優先審查資格的MEK抑制劑Cotellic® (cobimetinib, XL-518, GDC-0973, RG7420)合併BRAF抑制劑Zelboraf® (vemurafenib,2011核准)緊接著於2015年11月10日獲准上市,是由Roche藥廠所開發的組合藥物。Cotellic® 是Roche藥廠在過去5年中獲得U.S. FDA核准的第7個新藥,此藥物也在15天後獲得歐盟核准上市。U.S. FDA核准Cotellic® 是基於一項臨床III期試驗(coBRIM)結果,共包括495位先前未經治療但攜帶BRAF V600E突變的不可切除性局部晚期或轉移性黑色素瘤患者。數據顯示,Cotellic® 與Zelboraf® 合併使用時,客觀緩解率(objective response rate, ORR)為70%,PFS為12.3個月,平均總存活期(overall survival, OS)為22.3個月;單獨使用Zelboraf® 時,ORR為50%,PFS為7.2個月,OS為17.4個月(比化療延長3 個月)。此臨床結果亦證明同時抑制RAF和MEK比單獨抑制RAF延長5個月PFS;此外,使用Cotellic® /Zelboraf® 合併治療組1年存活率高達74.5%,2年存活率高達48.3%。
2011年以前,BRAF突變陽性晚期黑色素瘤患者只能以化療治療,客觀緩解率僅10%左右,總生存期不過數月而已,孰知僅僅4年時間,晚期黑色素瘤的治療從幾乎無藥可用到現在RAF、MEK、PD-1、CTLA4抑制劑加上T-VEC,已進入百家爭鳴的局面。Tafinlar® /Mekinist® 1個月藥價為1.53萬美元,1年藥價為18.36萬美元;Cotellic® /Zelboraf® 1個月藥價為1.76萬美元,1年藥價為21.1萬美元,專家預測銷售峰值可達8億美元。
Cotellic® 是由Exelixis公司利用片段設計(fragment-based drug design)為技術平台而開發出的MEK抑制劑,於2006年12月與Genentech公司(2009年被Roche藥廠併購為旗下子公司)簽署共同開發計畫,Exelixis從Genentech取得0.25億美元的簽約授權金和里程碑金。在2007年1月,Cotellic® 完成試驗中新藥(Investigational New Drug, IND)申請後,Exelixis再獲得0.15億美元里程碑金,並且負責Cotellic® 所有開發費用到臨床I期結束。接著在2009年3月開始臨床II期時,Exelixis又獲得700萬美元里程碑金,並由Genentech接手後續所有臨床開發費用。Exelixis與Genentech共享Cotellic® 在美國上市後的所有權益,另外Exelixis可獲得非美國地區的上市銷售權利金。目前Roche藥廠正在試驗Cotellic® 與其他藥物(包括免疫療法)用於多種類型腫瘤的治療潛力,包括非小細胞肺癌、結腸癌、三陰乳腺癌和黑色素瘤。
(4) Lenvima® (lenvatinib)
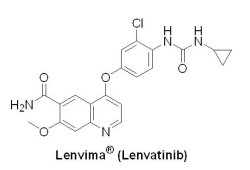 甲狀腺癌(thyroid cancer)是一種常見的內分泌腺惡性腫瘤,占全身惡性腫瘤的1%。南韓是全球發病率最高的國家,標準化發生率為67.9/10萬。根據衛生福利部國民健康署公布2012年新發生癌症人數,甲狀腺癌首次進入前十名,排名第九,新增人數為2,895人,標準化發生率9.9/10萬,高於全球平均發生率(4.0/10萬)。在2015年2月13日,U.S. FDA授予孤兒藥地位和優先審查資格的日本Eisai公司多靶點激酶抑制劑Lenvima® (lenvatinib, E7080)獲准上市,用以治療放射性碘復發之分化型甲狀腺癌(RR-DTC)患者。Lenvima® 是一種口服多受體酪氨酸激酶(RTK)抑制劑,具有新穎的結合模式,除抑制參與腫瘤增殖的其他促血管生成和致癌信號通路相關RTK(FGFR和RET)外,還能夠選擇性地抑制血管內皮生長因子(VEGFR2和VEGFR3)受體的激酶活性。
甲狀腺癌(thyroid cancer)是一種常見的內分泌腺惡性腫瘤,占全身惡性腫瘤的1%。南韓是全球發病率最高的國家,標準化發生率為67.9/10萬。根據衛生福利部國民健康署公布2012年新發生癌症人數,甲狀腺癌首次進入前十名,排名第九,新增人數為2,895人,標準化發生率9.9/10萬,高於全球平均發生率(4.0/10萬)。在2015年2月13日,U.S. FDA授予孤兒藥地位和優先審查資格的日本Eisai公司多靶點激酶抑制劑Lenvima® (lenvatinib, E7080)獲准上市,用以治療放射性碘復發之分化型甲狀腺癌(RR-DTC)患者。Lenvima® 是一種口服多受體酪氨酸激酶(RTK)抑制劑,具有新穎的結合模式,除抑制參與腫瘤增殖的其他促血管生成和致癌信號通路相關RTK(FGFR和RET)外,還能夠選擇性地抑制血管內皮生長因子(VEGFR2和VEGFR3)受體的激酶活性。Lenvima® 在2006年開始進行臨床I期試驗,2011年3月進入臨床III期試驗,U.S. FDA核准Lenvima® 是基於一項包括392位經先前放射碘治療後復發之分化型甲狀腺癌患者的臨床III期研究(SELECT)結果。資料顯示,口服Lenvima® (24毫克)治療組的PFS為18.3個月,ORR為65%;安慰劑組的PFS為3.6個月,ORR為2%。Lenvima® 在美國順利上市後,Eisai公司正全力推動Lenvima® 在全球超過20個國家上市,並積極開發Lenvima® 的其他適應症,例如肝細胞癌、腎細胞癌和非小細胞肺癌的治療,目標希望在2019年Lenvima® 年銷售峰值突破10億美元。根據Eisai公司全球癌症商業副總裁Ivan Cheung表示,Lenvima® 將是Eisai產品線最亮眼的皇冠寶石(the crown jewel of our portfolio)。
(5) Ibrance® (palbociclib)
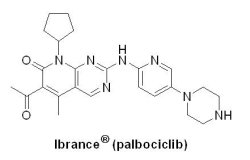 在2015年2月3日,美國Pfizer藥廠針對細胞週期素依賴性激酶CDKs 4/6生長訊號的一種口服選擇性抑制劑Ibrance® (palbociclib, PD-0332991),獲得U.S. FDA以突破性藥物資格、優先審查資格、加速核准機制同意上市,比預定審查期限提前了1年。一般來說,轉移性晚期乳癌患者在使用第一線荷爾蒙治療約十個月後,幾乎都會出現抗藥性,轉而使用第二或第三線荷爾蒙治療後,疾病控制的時間還會更短,最終會因為抗藥性的不斷產生,而須改用毒性較大的化學療法。CDKs 4/6是細胞週期的關鍵調節因素,能夠觸發細胞週期進展,是荷爾蒙受體陽性乳癌非常重要的訊息傳遞途徑之調節因素,於荷爾蒙治療出現抗藥性時仍占有重要的角色。
在2015年2月3日,美國Pfizer藥廠針對細胞週期素依賴性激酶CDKs 4/6生長訊號的一種口服選擇性抑制劑Ibrance® (palbociclib, PD-0332991),獲得U.S. FDA以突破性藥物資格、優先審查資格、加速核准機制同意上市,比預定審查期限提前了1年。一般來說,轉移性晚期乳癌患者在使用第一線荷爾蒙治療約十個月後,幾乎都會出現抗藥性,轉而使用第二或第三線荷爾蒙治療後,疾病控制的時間還會更短,最終會因為抗藥性的不斷產生,而須改用毒性較大的化學療法。CDKs 4/6是細胞週期的關鍵調節因素,能夠觸發細胞週期進展,是荷爾蒙受體陽性乳癌非常重要的訊息傳遞途徑之調節因素,於荷爾蒙治療出現抗藥性時仍占有重要的角色。U.S. FDA加速核准Ibrance® (II期臨床試驗PALOMA-1)是基於165位受試者接受Ibrance® 和letrozole(商品名:Femara® ) 合併治療後,PFS為20.2個月,ORR為55.4%;單獨使用letrozole組,PFS為10.2個月,ORR為39.4%。Ibrance® 每日口服1次,劑量為125毫克,連續服用21天,停藥7天,28天為1周期。Ibrance® 1個月藥價為0.985萬美元,1年藥價為11.82萬美元,全球醫藥行業調研機構GlobalData調查指出,Ibrance® 將主導HR+乳腺癌市場,預估到2023年的銷售額將達到18.5億美元。
Ibrance® 屬first-in-class藥物,背後也有一個淒美又苦盡甘來的故事。在1990年末期, Warner-Lambert藥廠旗下的Parke-Davis研發實驗室與Onyx藥廠以50/50比例共同合作開發 Ibrance®,Parke-Davis負責藥物化學、藥物動力學與藥物代謝、動物確效和藥物安全性評估,而Onyx因擁有諸多特有體外酵素和細胞篩選技術,故在此雙邊合作上,提供所有體外生物篩選模式。在2000年時,Pfizer藥廠透過併購Warner-Lambert取得Ibrance® 後續臨床試驗之主導權,而Onyx藥廠則擁有產品上市後8%的權利金收入。
Pfizer藥廠於2002年選定Ibrance® 為候選發展藥物,並於2004年進入臨床發展,但是直到2010年1月才正式進入臨床I期試驗,原因何在?一說是Pfizer藥廠在透過併購後,擁有Agouron/Parke-Davis/Pfizer的癌症藥物產品線(Warner-Lambert於1999年以21億美金併購Agouron生技公司),因當時有與Ibrance® 類似的細胞週期抑制藥物,在類似產品之相互競爭以及資源投入之優先性的情況下,導致Ibrance® 並沒有在第一時間下獲得高層青睞;另一說是Pfizer當時有多項臨床III期癌症藥物產品(例如Sutent® 、Inlyta® 、Xalkori® 、Bosulif® ),公司需要專注資源在late-stage program,而Ibrance® 當時仍屬於候選發展藥物階段,故資源投入之優先性遠低於臨床III期癌症藥物產品。幸運的是,Ibrance® 當時在資源豐富的大藥廠手上,並沒有被變賣或束之高閣;最後,在Pfizer藥廠龐大資源重新投入後,僅花了5年時間,順利將Ibrance® 從臨床I期推至上市核准。從上述Ibrance® 故事,也再一次印證大公司在併購之後,組織與人事的異動,常常為藥物開發帶來不可預測的變數。
(6) Farydak® (panobinostat)
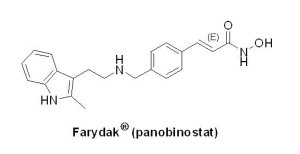 多發性骨髓瘤(multiple myeloma, MM)是一種復發率高和易產生抗藥性的疾病,其為一種源自於骨髓白細胞中漿細胞的惡性腫瘤。由於血漿內異常細胞的增殖導致腫瘤的發生,堆積在骨髓內使正常細胞被推擠並喪失正常功能。MM占所有癌症的1%,占血液系統癌症的10%,多發於60歲以上人群,全球約有11.4萬病例。2003年5月U.S. FDA核准Millennium Pharmaceuticals公司開發的Velcade® (bortezomib),為第一個用以治療MM的標靶藥物。隔了9年,才在2012年7月核准第二個以20S 蛋白酶體(proteasome)為標靶的藥物Kyprolis® (carfilzomib),是由Onyx藥廠所開發。又隔了3年,Novartis藥廠開發的first-in-class pan-HDAC抑制劑Farydak®(panobinostat, LBH-589)取得U.S. FDA授予孤兒藥地位、優先審查資格和加速核准資格,於2015年2月23日獲准上市用以治療MM。Farydak® 與Velcade® 和dexamethasone合併使用,用於先前至少接受2種治療方案(包括Velcade® 和一種免疫調節藥物)復發的MM患者。
多發性骨髓瘤(multiple myeloma, MM)是一種復發率高和易產生抗藥性的疾病,其為一種源自於骨髓白細胞中漿細胞的惡性腫瘤。由於血漿內異常細胞的增殖導致腫瘤的發生,堆積在骨髓內使正常細胞被推擠並喪失正常功能。MM占所有癌症的1%,占血液系統癌症的10%,多發於60歲以上人群,全球約有11.4萬病例。2003年5月U.S. FDA核准Millennium Pharmaceuticals公司開發的Velcade® (bortezomib),為第一個用以治療MM的標靶藥物。隔了9年,才在2012年7月核准第二個以20S 蛋白酶體(proteasome)為標靶的藥物Kyprolis® (carfilzomib),是由Onyx藥廠所開發。又隔了3年,Novartis藥廠開發的first-in-class pan-HDAC抑制劑Farydak®(panobinostat, LBH-589)取得U.S. FDA授予孤兒藥地位、優先審查資格和加速核准資格,於2015年2月23日獲准上市用以治療MM。Farydak® 與Velcade® 和dexamethasone合併使用,用於先前至少接受2種治療方案(包括Velcade® 和一種免疫調節藥物)復發的MM患者。值得一提的是,Farydak® 的核准之路可謂一波三折。2014年春天,Novartis藥廠提交新藥查驗登記(New drug application, NDA)的資料為一項包括768位試驗者之臨床III期研究(PANORAMA-1)結果,由於療效不錯,U.S. FDA遂同意將Farydak® 列為加速核准機制之案件,意即此審查週期將由通常的12個月縮短至8個月。然,2014年11月27日,U.S. FDA腫瘤藥物顧問委員會(Oncologic Drugs Advisory Committee, ODAC)以5比2的投票結果建議拒絕核准Farydak® ,主要理由是Farydak® /Velcade® /dexamethasone治療組的PFS雖可達3.9個月,但是當中有7%的患者死於與癌症無關的併發症,這些併發症包括了骨髓抑制、出血、感染、胃腸道毒性和心臟毒性等嚴重副作用,遠高於對照組的3.5%,故ODAC認為該藥物的副作用過於嚴重,要求U.S. FDA慎重考慮。接著,更大的挑戰是ODAC委員指出,Farydak® /Velcade® /dexamethasone治療組中有許多患者的PFS值因資料不完整和欠缺以致無法評估,並更進一步指出,若移除這些尚待釐清的Farydak® /Velcade® /dexamethasone治療組資料,其PFS僅比Velcade® /dexamethasone對照組延長了2.2個月,再加上當時Farydak® /Velcade® /dexamethasone治療組的次要終點,也就是整體存活率(OS)(33.6個月vs. 30.4個月)尚無法給予明確的結論(too early to make a call on overall survival)。ODAC的建議迫使U.S. FDA將原定於2014年12月的最後期限延遲至2015年3月。
Novartis藥廠於是修改了Farydak® 適應症並提交額外的分析資料,自原本768位試驗者之臨床III期資料中,重新分析193位先前曾接受Velcade® 和一種免疫調節藥物治療復發的MM患者於此試驗的治療結果,發現Farydak® /Velcade® /dexamethasone治療組的PFS為10.6個月,ORR為59%;而Velcade® /dexamethason對照組的PFS為5.8個月,ORR為41%,始獲得U.S. FDA同意核准上市,但須標示黑框警告,提示該藥物有嚴重的腹瀉及致命的心臟事件、心律失常及心電圖(ECG)變化。Farydak® 在美國21天的治療期程,需要花費6,860美金,預估銷售峰值達2.5億美元。
(7) Odomzo® (sonidegib)
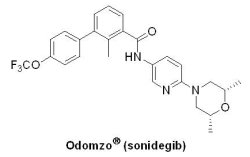 基底細胞癌(basal cell carcinoma, BCC)是一種最常見的皮膚癌,約占非黑色素瘤皮膚癌的80%以上,常病發於頭頸部。繼2012年U.S. FDA通過第一個由Roche藥廠所開發的Hedgehog訊號路徑抑制劑Erivedge® (vismodegib)後,在2015年7月24日通過第二個Hedgehog訊號路徑抑制劑,是由Novartis藥廠所開發的Odomzo® (sonidegib, erismodegib, LDE225, NVP-LDE225),用以治療經手術或放射治療後病情復發、及不適合上述2種治療方案的局部晚期基底細胞癌成人患者。
基底細胞癌(basal cell carcinoma, BCC)是一種最常見的皮膚癌,約占非黑色素瘤皮膚癌的80%以上,常病發於頭頸部。繼2012年U.S. FDA通過第一個由Roche藥廠所開發的Hedgehog訊號路徑抑制劑Erivedge® (vismodegib)後,在2015年7月24日通過第二個Hedgehog訊號路徑抑制劑,是由Novartis藥廠所開發的Odomzo® (sonidegib, erismodegib, LDE225, NVP-LDE225),用以治療經手術或放射治療後病情復發、及不適合上述2種治療方案的局部晚期基底細胞癌成人患者。U.S. FDA核准Odomzo® 是基於一項包括66位局部晚期基底細胞癌患者的臨床II期研究(BOLT)結果,於此評估了Odomzo® 200毫克和800毫克劑量的療效和安全性。結果顯示,口服200毫克劑量組的ORR為58%,其中完全緩解率(CR)為5%,部分緩解率(PR)為53%,有81%患者至少能夠維持1.9至18.6個月的疾病緩解。Odomzo® 目前正在進行多種癌症相關的臨床試驗開發,包括骨髓纖維化、白血病及實體瘤,如胰腺癌、乳腺癌和非小細胞肺癌。Odomzo® 1個月藥價約為1萬美元,預期銷售峰值達8億美元。
(8) Ninlaro® (ixazomib)
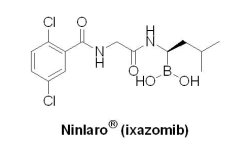 2015年可說是對抗MM疾病大豐收的一年,U.S. FDA共核准2個小分子藥物:Novartis藥廠開發的Farydak® (panobinostat)和Takeda公司開發的Ninlaro® (ixazomib, MLN9708),以及2個大分子單株抗體藥物:AbbVie與Bristol-Myers Squibb藥廠共同開發的EmplicitiTM(elotuzumab)和Johnson & Johnson公司的DarzalexTM(daratumumab),用於治療至少已接受一種標準治療方案而復發的MM患者。在2015年11月20日,U.S. FDA核准日本Takeda公司的Ninlaro® 上市,與Celgene公司的Revlimid® (lenalidomide)和dexamethasone(副腎皮質激素)共同治療MM患者。Ninlaro® 是繼2003年Velcade® (bortezomib)、2012年Kyprolis® (carfilzomib),第三個以蛋白酶體(20S proteasome)為藥物標靶上市的口服可逆抑制劑,主要是通過抑制多發性骨髓瘤細胞的酶活性,從而阻止癌細胞的生長和存活來發揮作用。有別於靜脈注射給藥或經由皮下注射的可逆抑制劑Velcade® 與靜脈注射給藥的不可逆抑制劑Kyprolis® ,Ninlaro® 是第一個口服的20S proteasome抑制劑(boronic ester prodrug),具孤兒藥地位(2020年11月20日)和優先審查資格,U.S. FDA是基於一項包括722位復發性多發性骨髓瘤患者參與的雙盲臨床研究結果核准Ninlaro® 上市。數據顯示,與安慰劑組相比(lenalidomide/dexamethasone),Ninlaro® /lenalidomide/dexamethasone治療組PFS為20.6個月,而安慰劑組PFS為14.7個月,效果顯著延長。
2015年可說是對抗MM疾病大豐收的一年,U.S. FDA共核准2個小分子藥物:Novartis藥廠開發的Farydak® (panobinostat)和Takeda公司開發的Ninlaro® (ixazomib, MLN9708),以及2個大分子單株抗體藥物:AbbVie與Bristol-Myers Squibb藥廠共同開發的EmplicitiTM(elotuzumab)和Johnson & Johnson公司的DarzalexTM(daratumumab),用於治療至少已接受一種標準治療方案而復發的MM患者。在2015年11月20日,U.S. FDA核准日本Takeda公司的Ninlaro® 上市,與Celgene公司的Revlimid® (lenalidomide)和dexamethasone(副腎皮質激素)共同治療MM患者。Ninlaro® 是繼2003年Velcade® (bortezomib)、2012年Kyprolis® (carfilzomib),第三個以蛋白酶體(20S proteasome)為藥物標靶上市的口服可逆抑制劑,主要是通過抑制多發性骨髓瘤細胞的酶活性,從而阻止癌細胞的生長和存活來發揮作用。有別於靜脈注射給藥或經由皮下注射的可逆抑制劑Velcade® 與靜脈注射給藥的不可逆抑制劑Kyprolis® ,Ninlaro® 是第一個口服的20S proteasome抑制劑(boronic ester prodrug),具孤兒藥地位(2020年11月20日)和優先審查資格,U.S. FDA是基於一項包括722位復發性多發性骨髓瘤患者參與的雙盲臨床研究結果核准Ninlaro® 上市。數據顯示,與安慰劑組相比(lenalidomide/dexamethasone),Ninlaro® /lenalidomide/dexamethasone治療組PFS為20.6個月,而安慰劑組PFS為14.7個月,效果顯著延長。Ninlaro® 和Velcade® 都是由Millennium Pharmaceuticals公司開發的20S proteasome抑制劑,日本Takeda公司是在2008年透過併購Millennium Pharmaceuticals公司取得這兩項藥物所有權。值得一提的是,Velcade® (bortezomib, MG-341, PS-341, LDP-341, MLM341, NSC-681239)在1995年即由MyoGenics公司開發出來(哈佛醫學院Alfred L. Goldberg教授為創辦人),後來該公司改名為ProScript,來由為proteasomes和transcription,故Velcade® 化合物專利(US 5,780,454;1998年7月14日)之專利所有權人為ProScript,之後由ProScript推展到臨床I期。由於ProScript在1999年6月23日被上市公司LeukoSite買下,4個月後(1999年10月18日),LeukoSite又被Millennium Pharmaceuticals公司以6.35億美元併購,而Millennium Pharmaceuticals公司在2008年5月14日再被日本Takeda公司以88億美元併購,之後Velcade® 就由Takeda與Johnson & Johnson公司共同開發,並於2003年5月13日上市(Takeda公司擁有美國市場,Johnson & Johnson公司擁有除美國以外的全球市場)。
由於Velcade® 在2014年的銷售額為29億美元,其物質專利將於2017年5月3日到期,但Takeda公司主張其延伸專利應到2022年,故在2012年Takeda公司控告Allergan公司所屬的Actavis、Novartis AG所屬的Sandoz和Accord Healthcare公司對於Velcade® 仿製藥之侵權訴訟。然而美國聯邦地區法院在2015年8月21日判Takeda敗訴,故Velcade® 專利維持在2017年5月到期。幸運的是,Takeda公司終於在Velcade® 到期一年多前,順利取得Ninlaro® 之上市。Ninlaro® 的起始推薦劑量為每週1次4毫克,分別在28天療程中的第1、8和15天口服給藥,價格為8,670美元。Ninlaro® 的口服給藥方式因方便患者的離院治療,因而更受MM患者的青睞;此外,Ninlaro® 的耐受性和安全性較好,與第一代蛋白酶體抑制劑Velcade® 相比,Ninlaro® 引起外周神經病變的風險更低。市場預估銷售峰值為15億美元。
(9) Lonsurf® (trifluridine/tipiracil)
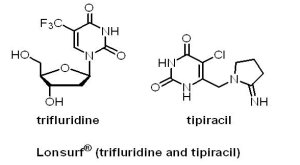 2015年9月22日,U.S. FDA核准日本Otsuka Holdings集團下Taiho Pharmaceutical公司的複方藥物Lonsurf® (trifluridine/tipiracil, FTD/TPI, TAS-102)上市,用於治療已接受2種治療方案復發之轉移性結直腸癌患者。Lonsurf® 是一種新型抗代謝複方藥物,是由具抗腫瘤活性的trifluridine(FTD)和胸苷磷酸化酶抑制劑(TPI)組成,分別有15毫克trifluridine/6.14毫克tipiracil和20毫克trifluridine/8.19毫克tipiracil。其中,FTD作用機制與5-FU類似,可在DNA複製過程中取代胸腺嘧啶直接嵌入DNA雙鏈,干擾癌細胞DNA的合成。FTD早在1964年就被Heidelberger等人合成出來,由於FTD口服吸收效果不好,經靜脈注射,其血中半衰期也只有12分鐘,故早期只用於眼睛感染皰疹病毒之治療。後來經過Taiho Oncology團隊研發,FTD在口服給藥後,很快就被thymidine phosphorylase(TP)酵素代謝成失去活性的代謝產物。接著,Taiho Oncology成功開發TPI tipiracil,能夠抑制與FTD分解相關的胸腺磷酸化酶,減少FTD的降解,維持FTD的血藥濃度。
2015年9月22日,U.S. FDA核准日本Otsuka Holdings集團下Taiho Pharmaceutical公司的複方藥物Lonsurf® (trifluridine/tipiracil, FTD/TPI, TAS-102)上市,用於治療已接受2種治療方案復發之轉移性結直腸癌患者。Lonsurf® 是一種新型抗代謝複方藥物,是由具抗腫瘤活性的trifluridine(FTD)和胸苷磷酸化酶抑制劑(TPI)組成,分別有15毫克trifluridine/6.14毫克tipiracil和20毫克trifluridine/8.19毫克tipiracil。其中,FTD作用機制與5-FU類似,可在DNA複製過程中取代胸腺嘧啶直接嵌入DNA雙鏈,干擾癌細胞DNA的合成。FTD早在1964年就被Heidelberger等人合成出來,由於FTD口服吸收效果不好,經靜脈注射,其血中半衰期也只有12分鐘,故早期只用於眼睛感染皰疹病毒之治療。後來經過Taiho Oncology團隊研發,FTD在口服給藥後,很快就被thymidine phosphorylase(TP)酵素代謝成失去活性的代謝產物。接著,Taiho Oncology成功開發TPI tipiracil,能夠抑制與FTD分解相關的胸腺磷酸化酶,減少FTD的降解,維持FTD的血藥濃度。Lonsurf® 是基於一項包括有800位已接受治療的轉移性結直腸癌患者參與的國際性、隨機、雙盲之臨床III期試驗(RECOURSE)結果獲得U.S. FDA核准上市。數據顯示,與安慰劑組相比,Lonsurf® 治療組OS為7.1個月,而安慰劑OS為5.3個月,同時Lonsurf® 治療組PFS為2個月,而安慰劑PFS為1.7個月。在此之前,Lonsurf® 已先於2014年3月在日本獲得核准;在歐洲,Taiho Pharmaceutical公司已于2015年3月向歐盟提交了Lonsurf® 的上市申請,並且於2015年6月15日與法國Servier簽署共同開發Lonsurf® 在歐洲的商業化銷售,Taiho Pharmaceutical公司可獲得簽約金1.3億美元以及未來授權金與銷售權利金。值得一提的是,Lonsurf® 是Taiho Oncology第一個獲得U.S. FDA核准之產品。
(10) Yondelis® (trabectedin)
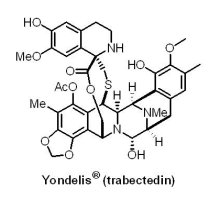 非常多的化療藥物是出自於天然物或其衍生物,早期有紫杉醇(Taxol® )、喜樹鹼(camptothecin)衍生物topotecan和irinotecan(CPT-11)、鬼臼毒素(podophyllotoxin)衍生物 etoposide、長春花鹼(Vinblastine® ),近期有埃博霉素B(epothilone B) 衍生物ixabepilone(Ixempra® )、抗生素rapamycin 衍生物everolimus(Afinitor® )、大田軟海綿素halichondrin B衍生物eribulin(Halaven® )。值得一提的是,eribulin是哈佛大學岸義人教授(Y. Kishi)於halichondrin B的全合成研究中所發現的。
非常多的化療藥物是出自於天然物或其衍生物,早期有紫杉醇(Taxol® )、喜樹鹼(camptothecin)衍生物topotecan和irinotecan(CPT-11)、鬼臼毒素(podophyllotoxin)衍生物 etoposide、長春花鹼(Vinblastine® ),近期有埃博霉素B(epothilone B) 衍生物ixabepilone(Ixempra® )、抗生素rapamycin 衍生物everolimus(Afinitor® )、大田軟海綿素halichondrin B衍生物eribulin(Halaven® )。值得一提的是,eribulin是哈佛大學岸義人教授(Y. Kishi)於halichondrin B的全合成研究中所發現的。在2015年10月23日,U.S. FDA通過優先審查機制,核准Johnson & Johnson公司旗下Janssen Pharmaceutical公司和西班牙Zeltia公司所開發的Yondelis® (trabectedin, ecteinascidin 743 or ET-743)上市,用於治療手術無法切除或轉移、曾接受過anthracycline化療治療的軟組織肉瘤(soft tissue sarcoma, STS),特別是脂肪肉瘤和平滑肌肉瘤患者。STS是一種起源於肌肉、脂肪、血管、神經、肌腱和關節內襯這類軟組織的腫瘤,占全部惡性腫瘤的0.7%,據統計,美國平均1年有12,000位STS患者。Yondelis® 屬於一種海洋生物鹼,主要的機制為藉由與腫瘤細胞雙股DNA 結合後(minor groove),在DNA鏈附近介入過氧化物的生產,生成Yondelis® -DNA產物,導致DNA骨架斷裂和細胞凋亡。Yondelis® 亦可抑制過度表現的多重抗藥性基因(multidrug resistance-1 gene, MDR-1),可以有效克服一般化療產生的抗藥性。Yondelis® 獲得U.S. FDA核准是基於一項包括518位轉移性脂肪肉瘤和平滑肌肉瘤患者之臨床III期試驗結果,其中有345位患者接受Yondelis® 治療,173位患者接受化療藥物dacarbazine治療;結果顯示,Yondelis® 治療組PFS為4.2個月,而對照組PFS則為1.5個月。
美國國家衛生研究院在1950至1960年建立一套大規模針對植物和海洋生物之抗癌活性篩選,而在1969年首次報導加勒比海鞘(Caribbean sea squirt)萃取物具有抑制癌細胞株生長之能力,但直到38年後,其效果才真正被意識到。其中,最具活性的化合物—海鞘素743(ET-743)之結構,在1984年才被美國伊利諾大學香檳分校的Kenneth Rinehart教授解出並申請專利;ET-743具有相當獨特的化學結構,含有3個四氫異喹啉環及一個含硫醚鍵的10環內酯。之後,伊利諾大學在1994年將其授權給西班牙PharmaMar公司(2015年6月30日Zeltia公司反向併購PharmaMar公司),並於1996年開始進行人體試驗。為了生產足夠的先導化合物ET-743用於臨床開發,PharmaMar公司養殖了250噸的Ecteinascidia turbinata(一種海鞘),然而,由於複雜的分離和純化過程導致ET-743的產量很低,每噸E. turbinata只能分離到近1克的產品,故Rinehart教授轉而與哈佛大學合成大師E. J. Corey教授(1990年諾貝爾化學獎得主)合作,在1996年開發出一個40多步的全合成方法,但是總產率低於2%。之後,PharmaMar公司透過生物發酵生產抗生素safracin B為前驅物,再透過不同保護基之最佳化,終於在2001年底,開發出半合成ET-743(18步)之放大製程,順利達到臨床III期試驗所需要的用量。在2001年8月,PharmaMar公司與Ortho Biotech Products(後併購為Centocor Ortho Biotech Products公司,再被Janseen Biotech公司併購,現成為Johnson & Johnson之子公司)簽訂共同開發ET-743合約,PharmaMar公司擁有歐洲市場,並獲得2,000萬美元之簽約金及不同階段里程碑金與10至20%權利金,而Ortho Biotech Products公司則擁有美國、日本和其他市場。值得一提的是,在與Ortho Biotech Products公司合作之前,PharmaMar公司也曾與Bristol-Myers Squibb(BMS)藥廠簽訂一項包含12個具抗癌效果的海洋生物評估案,但BMS最後並沒有同意此合作案。
有關臨床試驗部分,在Ortho Biotech Products公司接手推展美國市場之前,Yondelis® 已針對軟組織肉瘤、卵巢癌、乳癌、子宮內膜癌、前列腺癌和非小細胞肺癌在其他國家進行臨床試驗,並在2005年4月於卵巢癌治療方面取得U.S. FDA授予的孤兒藥地位。然而在2009年7月,ODAC評估包含672位復發性卵巢癌患者的臨床III期結果(OVA-301),認為Yondelis® 安全問題過大且療效並不顯著(治療組PFS為7.3個月 vs.對照組PFS為5.8個月),以14:1的投票結果建議拒絕核准Yondelis® 和Doxil® (liposomal doxorubicin)在復發性卵巢癌治療的聯合療法上市申請。有趣的是,當時菲律賓才剛剛通過Yondelis® 用於治療軟組織肉瘤,而歐洲早就在2007年就通過Yondelis® 用於治療卵巢癌。U.S. FDA遂要求Centocor Ortho Biotech Products公司提供進一步OVA-301患者之OS資料並重新進行臨床III期確效試驗。兩年後(2011年4月29日),Centocor Ortho Biotech Products公司同意自願放棄Yondelis® 在復發性卵巢癌治療的上市申請,並將Yondelis® 轉向惡性肉瘤的研發。終於Janssen Pharmaceutical藥廠在2014年11月24 日提出Yondelis® 在軟組織肉瘤之上市申請,在2015年2月3日通過優先審查機制後,於同年10月23日獲准上市。
Yondelis® 早在2007-2008年就已獲歐盟EMEA(2007年9月25日)、俄羅斯(2008年1月12日)和南韓(2008年5月19日)核准用於治療STS,2009年9月17日再下一城,獲得墨西哥、智利、烏拉圭、新加坡和越南的核准。Yondelis® 自1984年被分離鑑定出來,1996第一次進入臨床試驗,2009年第一次向U.S. FDA叩關失敗,持續6年與U.S. FDA搏鬥,歷經3次申請,終於在2015年在美國奪下聖盃;此外,透過日本合作夥伴Taiho Pharmaceutical公司的努力,順利地在2015年9月28日取得日本厚生勞動省的核准上市。Yondelis® 為針劑,一瓶藥價為2,700美元,每次需要3瓶,每21天給藥一次,目前在近80個國家上市銷售,全球年收入超過1億美元。
(11) Varubi® (rolapitant)
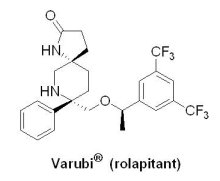 2015年9月2日,U.S. FDA核准總部在麻州Waltham市的Tesaro生技公司所開發的止吐藥Varubi® (rolapitant, SCH 619734)上市。Varubi® 可與其他抗嘔吐藥物聯合使用,用於防治癌症化療引發的噁心嘔吐之初始發作和反復發作。噁心嘔吐是接受化療治療的癌症患者中最常見的副作用,症狀可持續數天。在化療治療後24小時至120小時發生的噁心嘔吐,被稱為遲發性惡性嘔吐,易引發嚴重併發症。長期噁心嘔吐可能導致癌症患者體重下降、脫水、營養不良,嚴重影響患者的生活品質,使治療依從性明顯下降,甚至有些患者可能因此喪失生活的信心,選擇放棄治療。在預防化療所致噁心嘔吐(chemotherapy-induced nausea and vomiting, CINV)方面,目前有兩個類型的藥物,一類是5-HT3受體拮抗劑-Anzemet® (dolasetron)、Kytril® (granisetron)、Zofran® (ondansetron)及Aloxi® (palonosetron),另一類是神經激肽(Neurokinin 1, NK-1)受體拮抗劑-Emend® (aprepitant),而以NK-1受體拮抗劑效果較佳。自2003年Merck藥廠開發的高選擇性NK-1受體拮抗劑Emend® 獲准上市至今,尚未有第二個NK-1受體拮抗劑上市。在CINV癌症患者中,仍有高達一半的患者會經歷遲發性CINV(24至120小時),故CINV仍存在著未獲滿足的重大醫療需求。Varubi® 是P物質(substance P)/NK-1受體拮抗劑,血漿半衰期大約為7天,故對於遲發性CINV更有幫助。
2015年9月2日,U.S. FDA核准總部在麻州Waltham市的Tesaro生技公司所開發的止吐藥Varubi® (rolapitant, SCH 619734)上市。Varubi® 可與其他抗嘔吐藥物聯合使用,用於防治癌症化療引發的噁心嘔吐之初始發作和反復發作。噁心嘔吐是接受化療治療的癌症患者中最常見的副作用,症狀可持續數天。在化療治療後24小時至120小時發生的噁心嘔吐,被稱為遲發性惡性嘔吐,易引發嚴重併發症。長期噁心嘔吐可能導致癌症患者體重下降、脫水、營養不良,嚴重影響患者的生活品質,使治療依從性明顯下降,甚至有些患者可能因此喪失生活的信心,選擇放棄治療。在預防化療所致噁心嘔吐(chemotherapy-induced nausea and vomiting, CINV)方面,目前有兩個類型的藥物,一類是5-HT3受體拮抗劑-Anzemet® (dolasetron)、Kytril® (granisetron)、Zofran® (ondansetron)及Aloxi® (palonosetron),另一類是神經激肽(Neurokinin 1, NK-1)受體拮抗劑-Emend® (aprepitant),而以NK-1受體拮抗劑效果較佳。自2003年Merck藥廠開發的高選擇性NK-1受體拮抗劑Emend® 獲准上市至今,尚未有第二個NK-1受體拮抗劑上市。在CINV癌症患者中,仍有高達一半的患者會經歷遲發性CINV(24至120小時),故CINV仍存在著未獲滿足的重大醫療需求。Varubi® 是P物質(substance P)/NK-1受體拮抗劑,血漿半衰期大約為7天,故對於遲發性CINV更有幫助。U.S. FDA是基於3個隨機雙盲對照臨床III期的研究資料核准Varubi® 上市。在2,800位接受化療治療的癌症患者中進行試驗,治療組使用Varubi® /Kytril® /dexamethasone,對照組則是安慰劑/Kytril® /dexamethasone,資料顯示Varubi® 治療組在遲發期嘔吐次數、噁心及嘔吐急救藥物的使用明顯減少。
Rolapitant最早是由Schering-Plough公司研發,在2008年已完成3個臨床II期試驗。由於當時Schering-Plough公司和Merck藥廠正在著手洽談合併事宜,因Merck藥廠已擁有唯一上市的NK-1受體拮抗劑Emend® ,而Schering-Plough公司開發的rolapitant為新一代NK-1受體拮抗劑,為了避免聯邦貿易委員會(Federal Trade Commission)祭出反托拉斯法,而影響雙方合併事宜,故Schering-Plough急需為rolapitant尋找買主,最後在2009年10月13日以200萬美金權利金以及2,700萬未來里程碑金,將rolapitant轉讓給由Phillip Frost醫生創辦的OPKO Health公司;而Schering-Plough公司和Merck公司也在2009年11月4日順利完成合併。值得一提的是,Phillip Frost醫生在美國生技業可說是赫赫有名,他曾經在1972年創辦Key Pharmaceuticals公司,該公司後來在1986年被Schering-Plough以8.35億美金併購。1987年Frost醫生又與來自台灣的許照惠博士一起創辦Ivax Corporation,之後在2006年Ivax被以色列Teva Pharmaceutical公司以74億美金併購。Frost醫生自2007年擔任OPKO Health公司總裁至今,許照惠博士則擔任該公司副總裁兼技術長。
在2010年12月,Phillip Frost又將rolapitant轉賣給Lonnie Moulder於2010年創辦的Tesaro公司,其價值為1.21億里程碑金、20%銷售權利金和10% Tesaro的股票。主要的考量是Lonnie Moulder在任職MGI Pharma公司期間(2007年被日本Eisai公司以39億美元併購)有協助開發治療CINV藥物palonosetron(Aloxi® )之經驗,再加上Tesaro為新創公司,急需要有late-stage臨床藥物。在2013年12月23日,Tesaro公司宣布完成rolapitant兩個臨床III期試驗,結果顯示達到主要評估指標(primary endpoint),但並沒有達成次要評估指標(secondary endpoints),當天Tesaro股價大跌23%。之後,Tesaro公司再啟動第三個臨床III期試驗(532位試驗者),並在2014年9月8日向U.S. FDA提交3個臨床III期試驗結果的NDA申請,終於在2015年9月2日獲准上市。Varubi® 是Tesaro公司第一個上市產品,也是治療CINV的第二個NK-1受體拮抗劑。隨著Varubi® 正式登陸美國市場,也象徵著Merck藥廠止吐劑Emend® 獨霸CINV市場將面臨新勁敵。值得一提的是,在Varubi® 獲得U.S. FDA核准僅3週後,美國國家綜合癌症網絡(The National Comprehensive Cancer Network, NCCN)就將Varubi® 新增至《NCCN腫瘤學臨床實踐指南》作為一種推薦選擇。目前,Emend® 已銷往全球90多個國家和地區,年銷售額已超過5億美元,不過業界對Varubi® 十分看好,預計銷售峰值可達4億美元。由於Tesaro是一家小型生技公司(2010年創立,2012 IPO,市值為17.3億美元),要對上美國藥廠巨擘Merck(市值為1,513億美元),這場被喻為大衛王(David)與巨人歌利亞(Goliath)的小蝦米對抗大鯨魚戰爭,也正在美國上演中。
註1:漫談2014年FDA核准小分子新藥。《國家衛生研究院電子報−新藥大放異彩 第607、609、610期》,生技與藥物研究所謝興邦研究員、李靜琪副研究員。
註2:Morrison, C. Fresh from the biotech pipeline. Nature Biotech. 2016:34, 129-132.
註3:Novel Drugs 2015 Summary. www.fda.gov/drugs
註4:癌症相關故事已受邀刊登在中國化學會《化學》季刊7月號,2016。(尚未刊登)
《文/圖:生技與藥物研究所謝興邦研究員、李靜琪副研究員;審校:生技與藥物研究所陳炯東研究員》