NHRI Communications

會議報導
2009人體試驗研究人員講習班(二)
Clinical Investigator Training Class 2009 (Part II)
 四、知情同意的過程與受試者同意書
四、知情同意的過程與受試者同意書陳書毓督導長以其在實務操作上的經驗,說明知情同意過程與受試者同意書的關鍵要點。陳督導長指出,進行受試者知情同意流程時要確認:知情同意書是否獲IRB核准?過程中受試者是否是在實質逼迫的狀況下?受試者是否屬於弱勢族群(易受傷害族群)?是否有補簽同意書的狀況?細部的規範與定義可以參考美國PRIM&R(Public Responsibility in Medicine and Research)網站。
陳督導長表示,有效的知情同意過程必須包括3個要素:資訊揭露、合理解釋與理解、自願性同意,過程及同意書內容不宜過長,描述方式也要具有易讀性,且應著重在說明參加與不參加試驗的差別。陳督導長還強調,知情同意是一個持續進行的過程(圖二),從開始招募受試者到試驗結束都要持續關注及進行資訊揭露與修正後再同意的工作,甚至在某些試驗類型,研究完成後的一段時間內,都還可能與受試者有相關的互動,並不因取得同意書就完成知情同意的過程。
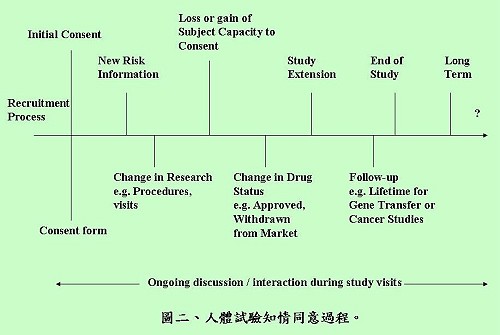
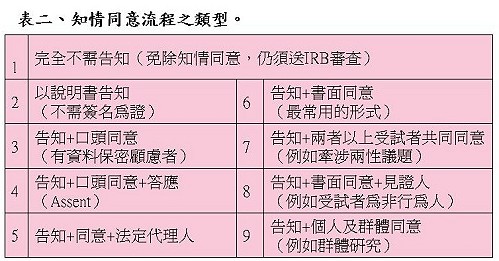
陳督導長提醒,知情同意書有許多類型,相關細節可以參考(彰化基督教醫院人體試驗委員會網站)及(美國Department of Health & Human Services)所列的同意書範本,依研究的性質不同,而有是否需告知、口頭同意及書面同意書之必要(表二)。
「可免除受試者同意書」之研究應該要同時具備4大要件:(1)研究對於受試者幾乎沒有風險,此處之風險包括生理、心理、社會、經濟上的風險;(2)免除同意書後對受試者的隱私與權益沒有不良影響;(3)不免除同意書則無法進行研究;(4)免除同意書後仍會適時提供受試者試驗相關訊息。然而針對可辨識的人進行研究,即使符合免除受試者同意書4大要件,還是需要預先進行IRB審查。如果僅是進行細菌分類或是細菌之安全性試驗,不特別需要進行人體試驗之研究,即不需預先進行IRB審查。如果是涉及動物試驗的研究,則必須準備書面資料送動物試驗委員會進行動物研究倫理審查。
五、研究人員對人體試驗委員會應有的認識
在此段課程,陳書毓督導長說明IRB的審查流程、修正案申請、嚴重不良反應事件(SAE)通報、及結案的相關規範,並佐以其參與審查的相關經驗,幫助學員瞭解相關規範設計的意義。
IRB主要的責任在保護受試者之權利、安全與福利。為求IRB審查時之公正客觀,其委員組成與比例也有一定規定。一般醫療機構接受評鑑時,評鑑項目之一即是IRB委員會委員組成是否符合9項組成規定,即:(1)具醫學背景之專業人員,例如醫師、藥師、護理師;(2)非醫學背景之專業人員,例如統計學家、倫理學家;(3)社會公正人士,例如社區里長或家庭主婦;(4)法律專家;(5)機構外人士;(6)男女比例,男或女的比例一定要超過1/3;(7)改聘(委員更換)比例也不能太高;(8)受試者(團體)代表;(9)諮詢專家,可彌補IRB專業委員對某些領域專業上的不足;這些委員的組成都要送衛生署報備。
IRB進行試驗倫理審查時必須依據基本的審查標準,陳督導長提出8項標準進行說明:(1)對受試者的風險應該是最小的;(2)風險與利益之比較應該合理;(3)受試者的選擇應該是公平合理的,也就是不要單挑某些族群做試驗,以免未來應用時對未受試驗族群造成安全性疑慮;(4)必須具備知情同意文件;(5)必須有適當的監測設計,要有資料安全監測計畫(DSMP)的規劃,有些風險性高的試驗還要成立監督小組(DSMB),定期提出監測報告,即時進行受試者保護;(6)確保受試者的安全性;(7)保護受試者的隱私;(8)嚴守資料保密的原則。
研究人員對於IRB的審查結果可以不認同,但是必須透過申覆或是列席說明的方式進一步溝通,而不能忽視IRB審查結果,逕自進行試驗。試驗進行過程中如果有需要修正之處,也必須提送相關資料給IRB進行審查,待IRB審查通過後才可進行變更之試驗,除非是需變更之試驗流程涉及嚴重緊急之安全性問題,則可在未獲核可狀況下,逕行試驗流程的變更,之後再向IRB報告。IRB除了進行試驗前及試驗修正之倫理審查之外,還要在試驗期間進行階段性(例如期中,不得少於每年一次)之追蹤審查,監督最初通過審查的試驗是否按照原來設計的流程與規範進行,確保受試者的權利與福祉。
對於不良事件的通報程序,陳督導長依據事件的輕重程度說明相對的通報機制(表三)。因各種狀況而至試驗執行結束時,試驗主持人除了向贊助者提交結案資料,還必須向IRB提出結案報告,即使試驗結果並未如預期。試驗主持人繳交的結案報告表中必須敘明收錄個案的描述、療效結果分析、不良事件結果分析。受試者同意書必須小心保管,同時還要繳交臨床試驗病人名單及成果報告。IRB則會針對受試者是否全部完成所有試驗、所有不良反應是否都獲得妥善處理、及主持人是否完成所有統計分析工作進行審查。
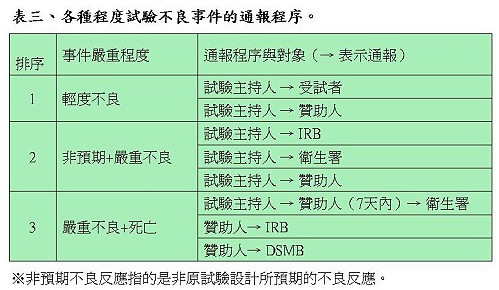
有關結案後所有試驗資料的保存,IRB必須至少保存三年,臨床試驗病例需永久保存,GCP第58條則規定贊助者對試驗資料至少要保存到藥品上市後2年。衛生署也針對試驗結果的公布原則進行規範,訂有「醫療機構及醫事人員發佈醫學新知或研究報告倫理守則」,供醫療機構及研究人員參考,避免不當方式及時間點公布過於誇大的試驗結果,誤導受試者或民眾而造成權益的損害。
六、藥品優良臨床試驗準則
課程最後由陳書毓督導長介紹藥品優良臨床試驗準則(GCP)各章節中有關試驗倫理的相關重點。GCP因為是針對藥品所進行的規範,所以試驗倫理的要求也比較嚴格。第一章總則定義所有相關的名詞,其中提到的「主持人手冊」內容含蓋試驗藥品之臨床及非臨床數據,是很重要的風險利益評估佐證。第二章說明受試者應獲得的保護與權益,以及保護的機制。第三章至第五章明確定義人體試驗委員會、試驗主持人及試驗委託者的角色與職責;第六章明列臨床試驗申請與審查要點,以及人體試驗委員會的追蹤審查機制;第七章明訂試驗過程中有關研究發現紀錄與報告的規範。
陳督導長最後提醒試驗主持人,無論是對於研究團隊或是受試者,都必須做整體有系統有效率的管理。在研究團隊部分,要時時磨練團隊管理的能力與技巧,要鼓勵工作人員提出問題,要傾聽研究護士的想法(瞭解試驗流程是否有問題),要建立團隊內部的一致性與協調機制。在受試者管理部分,要落實知情同意的流程,對受試者中途退出要坦然接受,對受試者要誠實對待。
重要參考資料
1. 2008年赫爾辛基宣言
2. 人體研究倫理政策指引
3. 臨床試驗受試者招募原則
4. 研究用人體檢體採集與使用注意事項
5. 98年5月20日醫療法最新修正條文
《文/圖:人體試驗研究人員講習班;編輯中心吳萃慧整理;審校:本院群體健康科學研究所衛生政策研究組許志成副研究員》