NHRI Communications

會議報導
疫苗臨床試驗案之要求與審查重點(一)
Conference report – Quality Concern in a Vaccine Clinical Trial (Part I)
 本院於2008年10月23日在竹南院區與財團法人醫藥品查驗中心及奇異亞洲醫療設備股份有限公司共同舉辦為期1天的「2008疫苗臨床試驗研討會」,針對疫苗臨床試驗之相關議題,邀請國內外專家學者前來進行經驗分享與意見交流,並紀念甫過世的抗癌鬥士朱夢麟醫師。會議中有關「疫苗臨床試驗案要求與審查重點」的議題,邀請查驗中心審查員李元鳳博士及詹明曉醫師分別就申請臨床試驗時,疫苗類藥品的品質、臨床要求、與審查重點進行說明。
本院於2008年10月23日在竹南院區與財團法人醫藥品查驗中心及奇異亞洲醫療設備股份有限公司共同舉辦為期1天的「2008疫苗臨床試驗研討會」,針對疫苗臨床試驗之相關議題,邀請國內外專家學者前來進行經驗分享與意見交流,並紀念甫過世的抗癌鬥士朱夢麟醫師。會議中有關「疫苗臨床試驗案要求與審查重點」的議題,邀請查驗中心審查員李元鳳博士及詹明曉醫師分別就申請臨床試驗時,疫苗類藥品的品質、臨床要求、與審查重點進行說明。疫苗類藥品的品質與審查重點
此場次中,李元鳳博士說明臨床試驗階段疫苗類藥品的化學、製造與管制 (Chemistry, Manufacturing and Controls, 以下簡稱 CMC)文件之重要項目及病毒安全控制注意事項。在第一期臨床試驗階段,優先考量的是疫苗的安全性相關品質管制,例如:不純物、無菌、熱原(pyrogen)、病毒安全性等。
申請第一期臨床試驗時,需要的技術性文件內容包括:(1)該疫苗的介紹(背景探討、欲測試或相似疫苗的人類使用經驗);(2)臨床研究流程(protocol),其中摘要及受試者同意書(Informed consent form,ICF)需要提供中文版本;(3)計畫主持人手冊(investigator brochure),內含CMC、動物藥理學研究及毒性試驗的結果等;(3)其他有助於該疫苗臨床試驗審查的相關資料。
第一期試驗疫苗提供的CMC文件內容包括:抗原及原料來源、外來物質(extraneous agents)的控制、製造流程、分析方法、疫苗成分組成及劑型、批次分析結果或檢驗成績書(Certificate of Analysis,COA),所收集的安定性資料至少足以支持疫苗在試驗期間的穩定性。李博士建議,樞鈕動物試驗與臨床試驗最好是使用同一批疫苗,如果無法使用同一批,則應提供不同批次疫苗之間品質相關的比較,此外,提供動物試驗比較的結果也會有幫助。
第二及第三期臨床試驗期間,額外蒐集的資料包括:(1) 批次分析結果,並與早期試驗的批次相比較,更新的安定性數據,製程及管控;(2) 製造流程的確效(validation)資料;(3) 分析方法的確效;(4)更新的原料藥及藥品規格(specification)等。
其中,製程及分析方法的確效是隨著臨床試驗的進程而進行(phase-in of validation),第一期臨床試驗的藥品會比較注重安全性相關的分析方法,例如宿主DNA、病毒PCR試驗等,皆宜有適當的確效試驗,例如靈敏度(sensitivity)及特異性(specificity)的建立。
疫苗的製造過程的病毒清除能力要求,會受到細胞株、病毒株、原料及製程中的病毒檢測的執行程度及其結果的影響,此外,病毒清除步驟的確效試驗,亦宜以模式病毒(model virus)來驗證疫苗的病毒清除的能力強度。
一般而言,製造第一期臨床試驗藥品的步驟,其累積的病毒清除(cumulative reduction)能力,如果是治療性藥品,宜至少達到開始病毒量的1/103,如果是用於健康受試者之預防用藥品則為1/106。此外,如果同一製造廠已有經由相同的製程製造的類似藥品的確效資料,於適當時可以做為審查的參考。
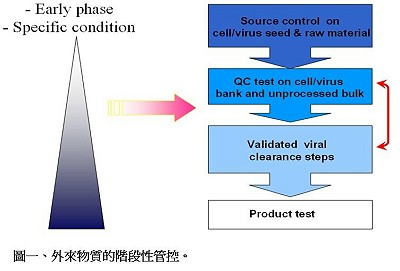
總而言之,包括病毒在內的外來物質的管控有數個層次(如圖一)。在試驗初期時,至少在細胞及病毒株的來源、原料的來源、細胞及病毒庫系統、粗製液(unprocessed bulk)階段應執行外來物質測試及管控;接下來則是病毒清除步驟的驗證;適當時,亦進行產品階段的測試。
演講者強調:本文論點為審查員對於疫苗類藥品的經驗分享,並不代表醫藥品查驗中心的立場
《文/圖:醫藥品查驗中心審查員李元鳳博士演講;編輯中心吳萃慧整理》