NHRI Communications

會議報導
醫療器材生技發展未來(二)
Local experts speak on future development of medical devices and biotechnology (Part 2 of 2)
 我國醫療器材管理之前瞻
我國醫療器材管理之前瞻黃小文博士負責衛生署藥政處之醫療器材法規規劃及國際合作。她表示,生技醫療器材(以下又稱醫材)產品上市前是需要被審查的,並分別從產品生命週期的各個階段(產品原型、臨床前測試、臨床試驗、量產、查驗登記、上市後不良反應通報、不良品回收等)進行管理,所以是一個門檻很高的領域,競爭性也高。過去10年,因為消費者保護的意識抬頭,各國醫藥審查管理制度也一直在變動,醫材產業所面臨的難題大多著重在風險管理的部分。國產醫材獲得我國上市許可證的比例很高,但是新醫材(例如塗藥支架、新診斷試劑)的審查較複雜,所需時間則較長。
我國醫療器材分級參照美國FDA制度,依據風險低至高簡單分為四種管理方式:第一等級(Class I)、第二等級(Class I)、第三等級(Class III)及新醫療器材(New,指台灣沒有類似品者)。第一級是風險最低的產品,可以臨櫃辦理許可證;高風險的醫材屬於第三級或新醫材者,風險高,技術性資料要求較多,審查標準也較高,審查時間一般較長。
黃博士表示,2006年我國醫藥器材國產品營業金額約占所有國產醫藥產品的47.9%(503.6億元新台幣),為我國醫藥產品主要外銷項目,進口者也約占所有進口醫藥產品的35%(379億元新台幣),2007年國內的GMP醫材廠商也增加至501家,營業額約有749億新台幣,目前仍持續成長中,顯示我國醫材產業仍積極發展中。為此,政府在2007年7月4日公告實施「生技新藥發展條例」,期望透過減稅措施,促進第三等級高風險高技術門檻醫材之商品化與企業投資,這個階段性政策截止日期為2021年12月31日,而這兩年間適法條件也逐步放鬆,有助於加速相關產業發展。
2009年所推動的生技起飛鑽石行動方案則以生技整合育成中心(Supra Incubator Center, SIC)園區發展模式,在台灣島北中南各區進行醫藥研發工作,期望與產業界進行緊密合作。北部為南港生醫園區,以生技製藥為主軸;中部為竹北生醫園區,以醫材為主軸;南部為路竹生醫園區,以牙科與整型醫材為主軸。此行動方案在高階醫材方面,從2009年起將從研發到產品上市分四階段扶植醫材產業:(1)基礎研發之臨床前測試國際標準採認;(2)轉譯研究之優良實驗室操作規範(GLP);(3)臨床試驗優良臨床試驗規範(GCP);(4)產品上市申請之優良製造規範(GMP)。黃博士認為,未來國家發展扶植這些生技產業時,最好能夠有效地將國家資源進行整合與串連,避免因為管理單位太多,使開發的技術片段化地進行,階段性計畫結束後無以為繼,既不利技術產品化,也不易與產業界銜接。
面對近年來醫療器材微小化、奈米化的進展,以及國際多元分工製造之趨勢,國外設計零件進口國內組裝的產品案件越來越多。另一方面,面對醫療器材與新興生物技術結合之趨勢,跨國性醫療器材臨床試驗的案件也逐漸增加,許多現有的審查經驗或法規標準與定義(藥事法第十三條、醫療器材管理辦法等)已經不足以因應日漸複雜的新興技術及其所可能衍生的權利義務關係與進行適當的安全風險評估,因此審查的挑戰度也越來越高。國內目前醫材審查工作由衛生署藥政處與藥檢局共同執行,預計2010年1月1日起由新成立的「台灣食品藥物管理局(TFDA)」之「醫療器材與化妝品組」負責管理。該組初分為6個科,與醫材有關之業務包括:(1)醫材安全及品質管理(負責不良反應Adverse Event通報及GMP查廠業務);(2)醫療器材基準;(3)新興醫療器材及臨床試驗管理;(4) 510k醫療器材(類似品,如同藥品類之學名藥)審查;(5)體外診斷醫療器材(例如診斷試劑)管理;(6)化妝品審查管理。此外,衛生署也正針對新興技術相關法規及審查標準的更新成立工作小組,參考歐美最新的標準,儘速研擬符合時宜並與國際接軌的法規。
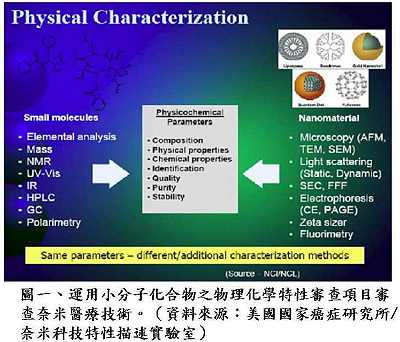 針對較特殊地輸入國外高科技技術並由國內製造組裝的新技術案件,如質子治療設備(proton therapy),衛生署因應的調整方案可由審查單位先評估國外零件的製造品管,認可其製造品質之後,廠商才能進口零件至國內進行組裝,組裝接近完成後要先評估最終成品在國內進行臨床試驗之成效,符合一切規範才能獲得上市許可。黃博士提醒,針對新興技術產品的審查,業者可以先與審查單位溝通,討論出以現有法規為主,並得以兼顧產業發展現況而予以變通的方式進行審查,例如奈米技術目前可以採用原有小分子化合物的審查原則加以因應(圖一),但最終還是要以藥物管理的目標為依歸,保障國人使用醫材的安全功效與品質。
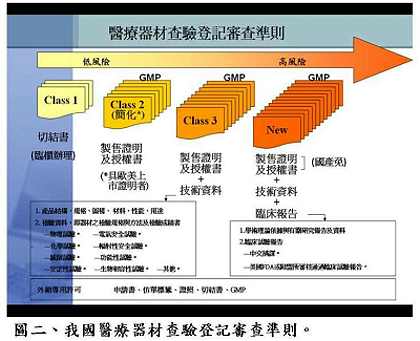
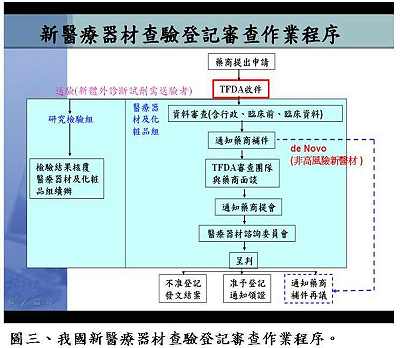
針對較特殊地輸入國外高科技技術並由國內製造組裝的新技術案件,如質子治療設備(proton therapy),衛生署因應的調整方案可由審查單位先評估國外零件的製造品管,認可其製造品質之後,廠商才能進口零件至國內進行組裝,組裝接近完成後要先評估最終成品在國內進行臨床試驗之成效,符合一切規範才能獲得上市許可。黃博士提醒,針對新興技術產品的審查,業者可以先與審查單位溝通,討論出以現有法規為主,並得以兼顧產業發展現況而予以變通的方式進行審查,例如奈米技術目前可以採用原有小分子化合物的審查原則加以因應(圖一),但最終還是要以藥物管理的目標為依歸,保障國人使用醫材的安全功效與品質。黃博士表示,國內醫療器材依據風險分類分別有各自的查驗登記審查準則(圖二),審查程序與美國食品藥物管理局十分類似,但多了廣告上的限制。審查時間則視審查產品的安全風險性而定,一般變更登記者約為30-60日,第二、三級者約為90-125天,通過率約為70%;新醫材則約要160天左右,通過率約為30%。未來配合TFDA之成立,新醫材查驗登記審查作業程序可能會有些微改變,對於非高風險性新醫材(De Novo Device),將考慮引入美國FDA之概念,簡化申請要求之資料(圖三),審查單位也會在把守安全、功效與品質之下,儘量加速審查。
由於各國對於醫材的風險等級認定標準不一,在美國可以視為第一等級的產品,在歐洲或日本不一定會被歸為相同之風險等級,因此企業對其產品也會考慮申請許可之難易度不同而有不同的國際上市佈局策略。此外,針對國外醫材來台申請許可證時,為求審慎,衛生署會要求產品應在原產國先上市,我國才會在審查通過後核發上市許可證,因此通常會與其原產國的上市許可時間晚一些,其間的時間差則要看產品的複雜度與風險性而定,從1個月至3年都是有可能的。此外,新的國際法規趨勢還強調原料供應商與進口商管理,避免因為上市後產品原料品質的不穩定,造成不良品或不良反應。
有關新醫療器材臨床試驗(一般稱為IDE,即Investigational Device Exempt)之審查程序,我國也參考美國FDA的作法,依照風險及用途,初期可以先進行小規模的可行性研究(feasibility studies)、繼以先驅試驗(pilot studies)或與核准與否之關鍵性臨床試驗(pivotal studies)。醫療器材臨床試驗的執行需要符合「醫療器材優良臨床試驗基準(GCP)」。臨床試驗用醫材應於衛生署及醫院倫理委員會(IRB)同意後始得進口並執行臨床試驗,醫材臨床試驗計畫案申請須知可參考衛生署相關網站。
黃博士表示,近年來衛生署也積極管理人體細胞組織衍生的醫材(human tissue derived device),並公告相關優良操作規範(Good Tissue Practice),內容包括進行捐贈者細胞組織篩檢追溯,避免交叉汙染;同時對於供查驗登記用地臨床前檢測資料,推動實驗室之優良實驗室操作準則(Good Laboratory Practice, GLP),進行GLP模擬查核與種子查核員訓練及GLP實驗室輔導,試行GLP自願性查核並發予「GLP自願性查核認可合格函」。此外,醫材核可上市後,衛生署會進行不良反應監測,透過醫材不良反應通報系統彙整並定期公告相關資訊。為協助醫材產業發展,衛生署特別提供「醫材產品開發相關法規諮詢服務窗口」,同時補助成立臨床試驗中心,建構高階醫材臨床試驗的環境。
衛生署也透過國際技術合作計畫(Technical Cooperation Programme)積極進行醫療器材國際合作,推動醫療器材優良製造操作(GMP)的品質系統文件(QSD)認證及產品QSD申請文件簡化模式,進行國際查廠及查廠報告資訊交流,使國際合作更具效益。現階段國際合作對象主要為美國FDA、歐盟12個代施查核單位(Notified Bodies)及瑞士合作,目前也正與澳洲藥物管理局(Therapeutic Goods Administration, TGA)洽談合作。面對亞太地區經濟逐漸茁壯,各國法規單位逐漸加強醫材管理,例如擁有極大市場的印度,又面對東南亞國協有ASEAN CSDT 2010醫療器材法規協定的現況,寄望我國醫材產業要加倍努力,爭取打入這個區域市場的先機。
《文:編輯中心吳萃慧整理;資料來源/圖:生技育成講座演講內容;審校:沈燕士董事長、黃小文博士》