NHRI Communications

知識產權
新藥大放異彩-漫談2014年FDA核准小分子新藥(下)「註1」
Introduction to FDA approved novel small molecular drugs in 2014 (part 3)
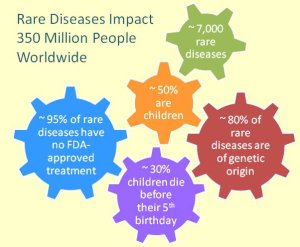 除了抗癌藥物、抗糖尿病藥物與感染性疾病藥物外,其他領域於2014年FDA核准小分子新藥部分,亦有不錯的表現,其中包括5個孤兒藥(orphan drug),分別是Cerdelga®、Esbriet®、Hetlioz®、NortheraTM和Ofev®。根據FDA的定義,「孤兒藥」是指疾病總人數低於20萬人的罕見疾病用藥,由於市場小且藥物開發需要大量經費與時間,因此藥廠在成本與利潤的考量下,大多不願意投入開發。為了鼓勵孤兒藥的研發,FDA於1983年首先制定《孤兒藥法案(Orphan Drug Act)》,並於1992年建立了加速審核程序,且提供研發租稅優惠、上市規費豁免、保障獨占市場地位及政府研發補助等優惠。現今,在大家日益重視罕見疾病患者的就醫權益與相關法規的優惠保障下,孤兒藥市場早已成為兵家必爭之地,不可同日而語。
除了抗癌藥物、抗糖尿病藥物與感染性疾病藥物外,其他領域於2014年FDA核准小分子新藥部分,亦有不錯的表現,其中包括5個孤兒藥(orphan drug),分別是Cerdelga®、Esbriet®、Hetlioz®、NortheraTM和Ofev®。根據FDA的定義,「孤兒藥」是指疾病總人數低於20萬人的罕見疾病用藥,由於市場小且藥物開發需要大量經費與時間,因此藥廠在成本與利潤的考量下,大多不願意投入開發。為了鼓勵孤兒藥的研發,FDA於1983年首先制定《孤兒藥法案(Orphan Drug Act)》,並於1992年建立了加速審核程序,且提供研發租稅優惠、上市規費豁免、保障獨占市場地位及政府研發補助等優惠。現今,在大家日益重視罕見疾病患者的就醫權益與相關法規的優惠保障下,孤兒藥市場早已成為兵家必爭之地,不可同日而語。其他領域
以下為13個其他領域藥物(Esbriet®、Ofev®、Striverdi® Respimat®、NeuracepTM、LumasonTM、Akynzeo®、MovantikTM、Cerdelga®、Hetlioz®、Belsomra®、ZontivityTM、Otezla®與NortheraTM)之相關開發過程:
(1) Esbriet® (pirfenidone)與Ofev® (nintedanib)
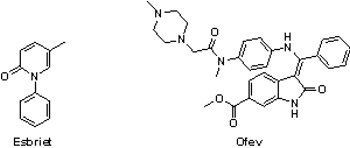 特發性肺纖維化(idiopathic pulmonary fibrosis, IPF)是一種持續發展、預後不佳的罕見疾病,目前發病機制尚未完全闡明。特發性肺纖維化患者肺部因未知的原因廣泛纖維化,因而失去交換氧氣的功能,導致最後死於呼吸衰竭,只有少數能經治療穩定病情或長期緩解。
特發性肺纖維化(idiopathic pulmonary fibrosis, IPF)是一種持續發展、預後不佳的罕見疾病,目前發病機制尚未完全闡明。特發性肺纖維化患者肺部因未知的原因廣泛纖維化,因而失去交換氧氣的功能,導致最後死於呼吸衰竭,只有少數能經治療穩定病情或長期緩解。2014年10月15日羅氏(Roche)公司之纖維化抑制劑Esbriet®(pirfenidone)與百靈佳殷格翰公司(Boehringer Ingelheim)之多蛋白激酶抑制劑Ofev®(nintedanib)同時獲FDA核准用以治療特發性肺纖維化,在預定的《處方藥使用者費用法案(Prescription Drug User Fee Act, PDUFA)》「註2」通過前,提早獲准上市(Esbriet®與Ofev®的《處方藥使用者費用法案》核准日期分別為2014年11月23日和2015年1月2日)。在此之前,特發性肺纖維化的治療,除了肺移植外就只有保守療法,FDA並未批准任何藥物用於此疾病的治療。
Esbriet®原是InterMune公司所研發的藥品,於2008年、2011年和2012年分別在日本、歐洲和加拿大獲准上市,然而因前3個臨床試驗的療效並不顯著,所以2010年FDA拒絕Esbriet®上市申請。不過,2014年的第4個臨床試驗結果證實Esbriet®可減緩患者肺功能的衰退,且達到6分鐘步行測試與無惡化存活時間(progression-free survival, PFS),終於獲得FDA的快速審核、優先審核、突破性藥物與孤兒藥等四重地位,於同年獲准上市。2014年8月24日Roche以每股74美元,總金額達83億美元收購InterMune,此為Roche自2009年以來最大宗的併購案,也助Roche強化在其治療呼吸系統疾病之領域。
Ofev®是一個針對血管內皮細胞生長因子受體(vascular endothelial growth factor receptor, VEGFR)、纖維母細胞生長因子受體(fibroblast growth factor receptor, FGFR)與血小板衍生生長因子受體(platelet-derived growth factor receptor, PDGFR)的多靶點激酶抑制劑,同樣獲得FDA的快速審核、優先審核、突破性藥物與孤兒藥等四重地位。Nintedanib亦可與歐洲紫杉醇(Docetaxel)合併使用治療肺腺癌患者,於2014年11月27日獲歐洲藥品管理局(European Medicine Agency, EMA)核准上市,商品名為Vargatef®。
特發性肺纖維化的藥物市場年銷售額預估將超過20億美元,此次FDA同時核准Esbriet®與Ofev®,勢必將為此市場帶來一場引人注目的商業競賽。只是,昂貴的藥價(Esbriet®每年94,000美元;Ofev®每年96,000美元)對患者而言,又是另一種負擔。
(2) Striverdi® Respimat® (olodaterol)
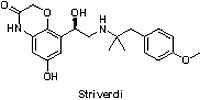 慢性阻塞性肺病(chronic obstructive pulmonary disease, COPD)是一種慢性發炎、無法完全治癒的呼吸道阻塞疾病,長期抽菸或吸入有害氣體是導致此疾病的主要原因。根據世界衛生組織(World Health Organization, WHO)的統計,2012年有超過300萬人死於慢性阻塞性肺病,相當於當年全世界所有死亡人數的6%。
慢性阻塞性肺病(chronic obstructive pulmonary disease, COPD)是一種慢性發炎、無法完全治癒的呼吸道阻塞疾病,長期抽菸或吸入有害氣體是導致此疾病的主要原因。根據世界衛生組織(World Health Organization, WHO)的統計,2012年有超過300萬人死於慢性阻塞性肺病,相當於當年全世界所有死亡人數的6%。2014年7月31日Boehringer Ingelheim公司所研發的Striverdi® Respimat®(olodaterol)獲FDA核准用於治療慢性阻塞性肺病,包括慢性支氣管炎和/或肺氣腫。Striverdi® Respimat®為具有高選擇性的吸入型長效β-腎上腺素受體促進劑(long acting β2-adrenergic agonist),其效果在超過4,900個確診為慢性阻塞性肺病受試者的臨床III期試驗中獲得證實:患者每天接受Striverdi® Respimat® (5 μg olodaterol)治療,經1秒用力呼氣量(forced expiratory volume in one second, FEV1)測試,結果顯示中度至極重度患者的肺功能獲得改善。
(3) NeuraceqTM(florbetaben F18 injection)與LumasonTM(sulfur hexafluoride lipid microsphere)
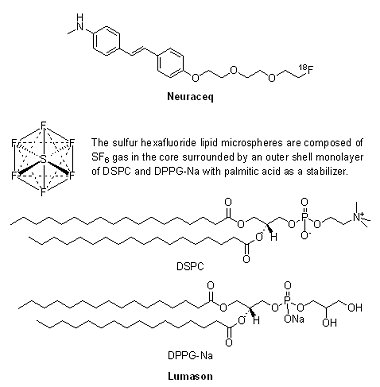 2014年有2個造影劑獲FDA核准上市,一為印度第八大製藥公司Piramal的NeuraceqTM(florbetaben F18 injection, BAY-949172),另一為Bracco Diagnostics公司的LumasonTM(SonoVueTM, sulfur hexafluoride lipid microsphere)。
2014年有2個造影劑獲FDA核准上市,一為印度第八大製藥公司Piramal的NeuraceqTM(florbetaben F18 injection, BAY-949172),另一為Bracco Diagnostics公司的LumasonTM(SonoVueTM, sulfur hexafluoride lipid microsphere)。2014年3月19日Piramal的NeuraceqTM獲FDA核准用於正子電腦斷層掃描(positron emission tomography, PET),利用β-澱粉樣蛋白斑(β-amyloid plaques)密度的PET成像,評估造成成人患者認知功能障礙的原因,例如是來自於阿茲海默症(Alzheimer’s Disease, AD)或是其他神經退化性疾病。NeuraceqTM是繼2012年的AmyvidTM(florbetapir F18 injection)和2013年的VizamylTM(flutemetamol F18 injection)第3個獲FDA核准的澱粉樣蛋白斑診斷試劑,經靜脈注射後,藉由[18F] Florbetaben與腦部的β-澱粉樣蛋白斑結合,產生能被PET檢測到的訊號。值得一提的是,NeuraceqTM開發權原屬於德國拜耳製藥(Bayer),Piramal公司於2012年4月15日與Bayer簽署協議,獲得Bayer分子影像研發之全球相關權利,其中包括阿茲海默症顯像劑florbetaben。
LumasonTM為超音波成像造影劑(ultrasound contrast agent),於2014年10月10日獲FDA核准用於利用超音波難以看到心臟超音波影像的患者。LumasonTM由位於美國紐澤西州Bracco Diagnostics上市銷售,此造影劑是藉由反射聲波來達到增強影像的效果,使醫師得以更清晰地看到患者心臟,其安全性與有效性在191個疑似心臟病患者參與的3個臨床試驗中被驗證。
(4) Akynzeo® (300 mg netupitant/0.5 mg palonosetron)
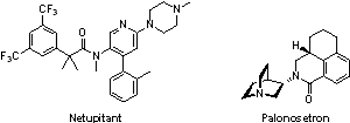 日本衛采公司(Eisai)所開發的Akynzeo®是由netupitant和Aloxi®(palonosetron)組成的口服複方藥物,於2014年10月10日獲FDA核准用以預防因化療所引起的噁心和嘔吐。Netupitant是神經激肽受體拮抗劑(neurokinin-1 (NK1) receptor antagonist),藉由阻斷神經激肽受體與物質P(substance P)的結合,可有效減緩噁心與嘔吐感,此藥物為用於預防癌症化療之急性期(化療開始24小時內)與延遲期(化療開始25-120小時內)所產生的噁心與嘔吐的一種新藥。Aloxi®則為具高度親合性的5-HT3受體拮抗劑(5-HT3 receptor antagonist),於2008年8月23日獲FDA核准用於預防癌症化療之急性期產生的噁心與嘔吐。Aloxi®和Akynzeo®原屬於瑞士Helsinn Healthcare擁有,於2010年6月10日由Eisai取得北美銷售權。
日本衛采公司(Eisai)所開發的Akynzeo®是由netupitant和Aloxi®(palonosetron)組成的口服複方藥物,於2014年10月10日獲FDA核准用以預防因化療所引起的噁心和嘔吐。Netupitant是神經激肽受體拮抗劑(neurokinin-1 (NK1) receptor antagonist),藉由阻斷神經激肽受體與物質P(substance P)的結合,可有效減緩噁心與嘔吐感,此藥物為用於預防癌症化療之急性期(化療開始24小時內)與延遲期(化療開始25-120小時內)所產生的噁心與嘔吐的一種新藥。Aloxi®則為具高度親合性的5-HT3受體拮抗劑(5-HT3 receptor antagonist),於2008年8月23日獲FDA核准用於預防癌症化療之急性期產生的噁心與嘔吐。Aloxi®和Akynzeo®原屬於瑞士Helsinn Healthcare擁有,於2010年6月10日由Eisai取得北美銷售權。相較於Aloxi®,Akynzeo®在1,720個接受癌症化療受試者參與的2個臨床試驗中證實能更有效預防因化療所引起的噁心和嘔吐。於第1個臨床試驗,服用Akynzeo®的受試者分別有98.5%、90.4%與89.6%在急性期、延遲期與整個化療療程皆無嘔吐或需額外藥物支持的現象發生,第2個臨床試驗的結果亦然。
(5) MovantikTM (naloxegol)
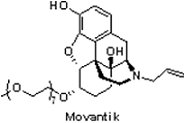 鴉片類藥物(Opioids)藉由與中樞神經系統(central nervous system, CNS)的μ-鴉片受體結合達到緩解慢性疼痛的效果,然而,其亦與胃腸道之μ-鴉片受體作用,因此導致鴉片誘導性便秘(opioid-induced constipation),而此常見的副作用通常會持續整個療程。
鴉片類藥物(Opioids)藉由與中樞神經系統(central nervous system, CNS)的μ-鴉片受體結合達到緩解慢性疼痛的效果,然而,其亦與胃腸道之μ-鴉片受體作用,因此導致鴉片誘導性便秘(opioid-induced constipation),而此常見的副作用通常會持續整個療程。2014年9月16日FDA核准AstraZeneca公司與Nektar Therapeutics公司聯合開發的MovantikTM(naloxegol),用於緩解慢性非癌症成人患者因服用鴉片類藥物所引起的便秘。值得注意的是,Salix製藥原本僅用於治療安寧照護下重症患者因鴉片類止痛藥引起的便秘症狀的Relistor® (methylnaltrexone bromide),亦幾乎同時(2014年9月29日)獲FDA核准將治療對象擴展到非癌症的慢性病患者。MovantikTM與Relistor®均屬於周邊作用的μ-鴉片受體拮抗劑(peripherally acting μ-opioid receptor antagonist),亦適用於相同的治療領域;然而,MovantikTM每日口服1次的獨特優勢將使需皮下注射的Relistor®難以與之抗衡。
MovantikTM在2個為期12週、共1,352名因服用鴉片類止痛藥而引起便秘症狀的非癌症慢性病患者參與的臨床試驗中,被證實確能顯著提高緩解比例。於第1個臨床試驗中,每天服用1次12.5毫克或25毫克MovantikTM的受試者,分別有41%和44%達到每週腸蠕動次數增加的效果,第2個臨床試驗亦有類似的結果。於安全性方面,FDA要求待MovantikTM上市後,需進一步評估服用此藥物的患者是否有心血管潛在風險。
MovantikTM為Nektar Therapeutics利用其口服小分子聚合物共軛技術所研發之藥物(PEGylated naloxol),於2009年9月21日,AstraZeneca與Nektar Therapeutics簽署合作協議,由AstraZeneca負責MovantikTM的開發和商業化,且根據後續修正之協議,AstraZeneca需在FDA接受MovantikTM申請的5天內,支付Nektar Therapeutics 7,000萬美元里程碑款項;目前Nektar Therapeutics已獲得此款項。此外,AstraZeneca於2015年3月19日宣布將與日本第一三共(Daiichi Sankyo)共同在美國推銷MovantikTM,Daiichi Sankyo將支付6.25億美元作為銷售支出,而AstraZeneca則負責所有的生產與銷售,並支付Daiichi Sankyo MovantikTM的銷售佣金。
(6) Cerdelga® (eliglustat)
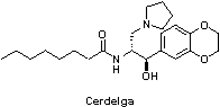 高雪氏症(Gaucher disease)為染色體隱性遺傳疾病,是溶酶體貯積病(lysosomal storage disease)中最常見的一種。高雪氏症是一種罕見疾病,目前全球約僅有1萬名患者,主要是因患者體內葡萄糖腦苷酯酶(gIucocerebrosidase)基因突變,造成單核巨噬細胞內的葡萄糖腦苷脂無法被水解而堆積在溶酶體中形成高雪氏細胞,進而導致肝脾腫大、貧血與骨骼發展等病變。高雪氏症依據發病的緩急、內臟受累程度以及有無神經系統症狀,可分成I型(非神經型、成人型、慢性型)、II型(急性型、神經型)和III型(亞急性型、神經型),其中以I型最常見。
高雪氏症(Gaucher disease)為染色體隱性遺傳疾病,是溶酶體貯積病(lysosomal storage disease)中最常見的一種。高雪氏症是一種罕見疾病,目前全球約僅有1萬名患者,主要是因患者體內葡萄糖腦苷酯酶(gIucocerebrosidase)基因突變,造成單核巨噬細胞內的葡萄糖腦苷脂無法被水解而堆積在溶酶體中形成高雪氏細胞,進而導致肝脾腫大、貧血與骨骼發展等病變。高雪氏症依據發病的緩急、內臟受累程度以及有無神經系統症狀,可分成I型(非神經型、成人型、慢性型)、II型(急性型、神經型)和III型(亞急性型、神經型),其中以I型最常見。自1991年以來,酵素替代療法(enzyme replacement therapy)已成為高雪氏症的標準治療方法,患者需定期透過靜脈注射施打藥劑(每兩週1次),因此當賽諾菲(Sanofi)公司透過併購健贊(Genzyme)取得歷經15年、遍及29個國家、約400名患者參與的Cerdelga®(eliglustat)於2014年8月19日獲FDA核准,成為用於I型高雪氏症成人患者唯一的口服用藥,著實為當前注射型藥物的高雪氏症市場帶來巨大的衝擊。Cerdelga®的年銷售額預估到2020年將達到7.49億美元。
有別於酵素替代療法降解沉積在細胞中的脂肪沉積物的機制,Cerdelga®則是直接抑制脂肪沉積物堆積在細胞中。159名接受過酵素替代療法穩定的I型高雪氏症患者分別使用酵素替代藥物Cerezyme®(imiglucerase for injection)與Cerdelga®,結果顯示二者對於穩定血紅素濃度、改善血小板計數及肝脾體積之效果相當。此外,於40名未接受過酵素替代治療的I型高雪氏症患者的臨床試驗中,受試者每天服用兩次42毫克Cerdelga®,治療4週之後,改為每天服用兩次84毫克的Cerdelga®,結果顯示受試者的脾臟體積在第39週時有顯著地縮小,且對肝臟體積、血小板計數及血紅素濃度亦有顯著地改善。
(7) Hetlioz® (tasimelteon)與Belsomra® (Suvorexant)
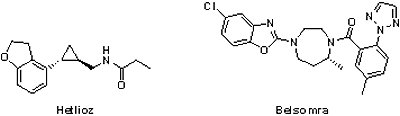 2014年共有2個治療睡眠困擾之藥物獲FDA核准上市,一為Vanda Pharmaceuticals公司的Hetlioz®(tasimelteon, VEC-162, BMS-214,778),另一為Merck公司開發的Belsomra®(Suvorexant, MK-4305);前者是針對非24小時睡眠-清醒障礙(non-24-hour sleep-wake disorder)的治療,後者則為一般失眠用藥。
2014年共有2個治療睡眠困擾之藥物獲FDA核准上市,一為Vanda Pharmaceuticals公司的Hetlioz®(tasimelteon, VEC-162, BMS-214,778),另一為Merck公司開發的Belsomra®(Suvorexant, MK-4305);前者是針對非24小時睡眠-清醒障礙(non-24-hour sleep-wake disorder)的治療,後者則為一般失眠用藥。2014年1月31日獲FDA核准用於治療非24小時睡眠-清醒障礙的Hetlioz®是一個孤兒藥,也是第一個獲FDA核准用於此領域的藥物。非24小時睡眠-清醒障礙主要發生在盲人身上,由於看不見陽光,所以他們的生物時鐘往往長於24小時,因而與正常周期(24小時)產生時差,導致他們經常在夜間失眠而白天疲倦,目前美國約有10萬盲人有此困擾。Hetlioz®是褪黑激素受體促進劑(melatonin receptor agonist),其有效性已於2個臨床試驗共104個盲人受試者身上獲得證實。相較於安慰劑組,服用Hetlioz®的受試者在增加夜間睡眠與降低白天睡眠的持續時間均有顯著的改善,只是Hetlioz®會降低使用者的警覺性,因此需在睡前固定時間服用,且應限制服藥後的活動。Hetlioz®原開發廠商為必治妥施貴寶(Bristol-Myers Squibb),Vanda Pharmaceuticals於2004年2月透過專屬授權取得,包括50萬先期技轉金、4,000萬的里程碑金和上市後權利金。Vanda Pharmaceuticals因Hetlioz®的開發獲美國罕見疾病組織(National Organization for Rare Disorders)頒發「2015年企業創意獎(Industry Innovation Award)」。
2014年8月13日獲准上市的Belsomra®是一具新穎機制市場首見(first-in-class)的安眠藥,不同於傳統安眠藥利用活化能夠舒緩焦慮的神經傳導物質來治療失眠,Belsomra®為食慾素受體拮抗劑(orexin receptor antagonist),主要是藉由抑制保持清醒的神經傳導物質食慾素,達到促進並維持睡眠的作用,其有效性已在超過500個受試者的3個臨床III期試驗結果獲得證實,結果顯示Belsomra®可幫助患者快速入睡並減少半夜醒來的時間。然而,由於考量服用最佳劑量30毫克或40毫克Belsomra®會使患者增加第二天嗜睡的風險,因此FDA曾在2013年7月拒絕核准Belsomra®的新藥申請,此次核准僅限於低劑量的使用(5毫克、10毫克、15毫克和20毫克),並建議醫師自最小劑量開始給藥,同時需告誡接受20毫克劑量之患者隔天禁止開車,主要是因為Merck追蹤服用20毫克Belsomra®的患者第二天的駕駛情況,結果發現不論男女的駕駛能力均受到影響。目前失眠市場已充斥相當多學名藥,包括最新暢銷藥物Stilnox®(zolpidem,史蒂諾斯)的仿製藥,故在這種情況下,該如何說服美國6,000萬失眠患者嘗試使用僅基於安慰劑對照研究,而非與其他失眠藥物比較的研究即獲核准上市的Belsomra®,對Merck而言,可謂是一場艱苦的戰鬥。
(8) ZontivityTM (vorapaxar)
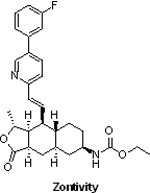 2014年5月8日FDA核准由Merck所研發之抗凝血劑ZontivityTM (vorapaxar,MK-5348,SCH 530348),此藥物為市場首見的蛋白酶激活受體1(protease-activated receptor 1, PAR-1)拮抗劑,可以抑制血小板聚集,從而減少血液凝塊的形成,用以降低有心肌梗塞或末梢血管疾病史的患者之心臟病發作、中風、心血管疾病死亡與血管重建等風險。
2014年5月8日FDA核准由Merck所研發之抗凝血劑ZontivityTM (vorapaxar,MK-5348,SCH 530348),此藥物為市場首見的蛋白酶激活受體1(protease-activated receptor 1, PAR-1)拮抗劑,可以抑制血小板聚集,從而減少血液凝塊的形成,用以降低有心肌梗塞或末梢血管疾病史的患者之心臟病發作、中風、心血管疾病死亡與血管重建等風險。由有26,449例患者的臨床試驗(Thrombin Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events)結果顯示,在3年間,ZontivityTM與其他抗血小板藥物(Aspirin® 與/或Plavix®)合併使用,可使患者心肌梗塞、中風、心血管疾病死亡的風險自9.5%降至7.9%,此顯著效果使ZontivityTM獲FDA顧問委員會以10:1的投票結果核准上市。
(9) Otezla® (apremilast)
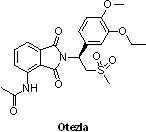 賽基公司(Celgene)所研發之Otezla®(apremilast,CC-10004)為市場首見的口服磷酸二酯酶第4型(phosphodiesterase 4)選擇性抑制劑,於2014年3月21日獲FDA核准用於治療活動性乾癬性關節炎(active psoriatic arthritis)成人患者,並於2014年9月25日獲准用於中重度斑塊型乾癬症(plaque psoriasis)的治療。
賽基公司(Celgene)所研發之Otezla®(apremilast,CC-10004)為市場首見的口服磷酸二酯酶第4型(phosphodiesterase 4)選擇性抑制劑,於2014年3月21日獲FDA核准用於治療活動性乾癬性關節炎(active psoriatic arthritis)成人患者,並於2014年9月25日獲准用於中重度斑塊型乾癬症(plaque psoriasis)的治療。乾癬性關節炎是一種慢性發炎疾病,據估計,全世界大約有3,800萬人罹患此疾病,而Otezla®是目前唯一獲得FDA核准的乾癬性關節炎口服治療藥物。乾癬性關節炎臨床療效之長期評估(Psoriatic Arthritis Long-term Assessment of Clinical Efficacy, PALACE)PALACE I、II與III的臨床試驗結果顯示,乾癬性關節炎成人患者在接受Otezla®治療16週後,達到ACR20(American College of Rheumatology 20,美國風濕科學會20%改善標準)、ACR 50和ACR 70改善標準的患者比例均顯著增加,疾病相關的身體功能也有改善。
乾癬(牛皮癬,銀屑症)是免疫反應所引起的慢性皮膚疾病,目前全球患者人數已超過1.25億人。兩個Otezla®對乾癬患者的臨床療效與安全性之評估(Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis, ESTEEM)ESTEEM I和ESTEEM II結果顯示,中重度斑塊型乾癬患者接受Otezla®治療後,斑塊狀乾癬體表面積不斷縮小,對頭皮、指甲、手掌和腳掌等難治部位亦呈現改善效果。此外,Otezla®治療中度至重度斑塊型乾癬的療效足以媲美安進(Amgen)的生物製劑Enbrel®(etanercept,2013年銷售額達83億美元)。
(10) NortheraTM (droxidopa)
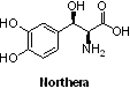 2014年2月18日NortheraTM(droxidopa,SM-5688)獲FDA加速核准用於治療原發性自主神經衰弱、多巴胺β羥化酶缺乏症與非糖尿病自主神經病變等患者之神經源性姿態性低血壓(neurogenic orthostatic hypotension),是一種孤兒藥。NortheraTM原始開發商為日本住友製藥(Sumitomo Pharmaceuticals),1989年已在日本上市;2005年10月1日大日本製藥(Dainippon Pharmaceuticals)與Sumitomo Pharmaceuticals合併成為日本第6大藥廠-大日本住友製藥(Dainippon Sumitomo Pharma),而Chelsea Therapeutics公司則於2006年5月30日自Dainippon Sumitomo Pharma取得除日本、韓國、中國和台灣以外之全球開發權。也因為NortheraTM在2014年順利上市,丹麥第2大製藥公司H. Lundbeck A/S於2014年5月8日遂以6.45億美金併購Chelsea Therapeutics,已取得NortheraTM未來銷售權。NortheraTM的年銷售額預估約為3至3.75億美元。
2014年2月18日NortheraTM(droxidopa,SM-5688)獲FDA加速核准用於治療原發性自主神經衰弱、多巴胺β羥化酶缺乏症與非糖尿病自主神經病變等患者之神經源性姿態性低血壓(neurogenic orthostatic hypotension),是一種孤兒藥。NortheraTM原始開發商為日本住友製藥(Sumitomo Pharmaceuticals),1989年已在日本上市;2005年10月1日大日本製藥(Dainippon Pharmaceuticals)與Sumitomo Pharmaceuticals合併成為日本第6大藥廠-大日本住友製藥(Dainippon Sumitomo Pharma),而Chelsea Therapeutics公司則於2006年5月30日自Dainippon Sumitomo Pharma取得除日本、韓國、中國和台灣以外之全球開發權。也因為NortheraTM在2014年順利上市,丹麥第2大製藥公司H. Lundbeck A/S於2014年5月8日遂以6.45億美金併購Chelsea Therapeutics,已取得NortheraTM未來銷售權。NortheraTM的年銷售額預估約為3至3.75億美元。NortheraTM是目前唯一獲FDA核准用於神經源性姿態性低血壓治療的藥物。神經源性姿態性低血壓是一種罕見的慢性姿態性低血壓,其與帕金森氏症、多系統萎縮症及單純性自主神經衰弱有關,患者常因頭暈、視力模糊、無力、注意力不集中及站立時昏厥等原因嚴重影響日常活動的能力。於兩項周期為2週的臨床試驗結果顯示,神經源性體位性低血壓患者在服用NortheraTM後,可有效降低頭暈目眩、感覺模糊或彷彿暫時失去知覺的現象,只是症狀改善超過2週的持久性尚未得到證實。
2014年是新藥大放異彩的一年,顯示FDA在審查效能與態度上的提升,相對的,也提振了相關產業的信心。不可否認,從上、中、下3集之瞭解,幾乎每一項藥物都有其辛酸史,不論是臨床進展不順利,甚或新藥核准申請被拒後捲土重來。另外,從核准的新藥中不難發現,有65%核准新藥是透過併購或技轉取得,公司的併購交易似乎已成了新趨勢,2014年製藥業的併購交易金額達到2,500億美元,創了有史以來的紀錄。此外,罕見疾病藥物與生物藥的研發亦是此波新藥中備受矚目的一環,前者在相關法規的優惠保障下獲得製藥業的青睞,為罕見疾病患者的治療帶來新契機;後者自過去數年來,在全球市占率呈現逐年攀升的趨勢。2014年FDA除了核准30個小分子以外,亦核准了11個生物藥,其相關訊息將於近期介紹。
註1:新藥大放異彩-漫談2014年FDA核准小分子新藥的報導,主要是根據註3至註5這3篇文章所提供之資訊撰寫。
註2:二十多年前,一款新藥若要通過FDA的審查,從遞交申請到核准上市平均需花費30個月,相當冗長耗時。然而,美國國會於1992年通過《處方藥使用者費用法案(Prescription Drug User Fee Act, PDUFA)》,送審業者必須支付審查費用給FDA,這筆「使用者費用」可以讓FDA聘僱更多專業人士,不僅加快審核的速度,也提升專業水準。《處方藥使用者費用法案》最近一次的更新在2012年,目前新藥從審核到上市平均只需15個月,而治療某些疾病的藥物如愛滋病,更能優先審核,平均只需6個月。
註3:Morrison, C. Fresh from the biotech pipeline-2014. Nature Biotechnology. 2015, 33, 125-128.
註4:Jarvis, L. M. The year in new drugs. Chemical & Engineering News. 2015 February 2, 11-16.
註5:Mullard, A. 2014 FDA drug approvals. Nature Reviews Drug Discovery. 2015, 14, 77-81.
新藥大放異彩-漫談2014年FDA核准小分子新藥(上)。《國家衛生研究院電子報607期》。
新藥大放異彩-漫談2014年FDA核准小分子新藥(中)。《國家衛生研究院電子報609期》。
《文/圖:生技與藥物研究所謝興邦研究員、李靜琪副研究員;審校:生技與藥物研究所陳炯東研究員》