NHRI Communications

研究發展
自動化次世代定序分析軟體將促進找尋家族性的罕見致病基因突變
NHRI researcher develops an automatic next-generation sequencing analysis pipeline to facilitate the identification of rare variants associated with diseases in families
 次世代定序技術已成為找尋罕見致病基因突變的利器,藉由全外顯子(whole exome)甚至全基因體(whole genome)的定序,可找出患者染色體上的突變;而透過家族資料,可找出患病親屬間共有的基因突變。然而,分析次世代定序資料需要結合各種生物資訊工具、統計分析軟體及外部資料,如人類基因組參考序列資料、序列比對生物資訊軟體及連鎖分析統計分析軟體等,增加了分析上的困難度。為了簡化及加速次世代定序資料的分析,目前已有許多自動化次世代定序資料分析的流程軟體,但這些軟體功能多注重在上游的序列分析如基因型的決定等,在決定基因型後,如何快速找到患病親屬間共有的基因突變,仍是分析次世代定序家族資料的挑戰。
次世代定序技術已成為找尋罕見致病基因突變的利器,藉由全外顯子(whole exome)甚至全基因體(whole genome)的定序,可找出患者染色體上的突變;而透過家族資料,可找出患病親屬間共有的基因突變。然而,分析次世代定序資料需要結合各種生物資訊工具、統計分析軟體及外部資料,如人類基因組參考序列資料、序列比對生物資訊軟體及連鎖分析統計分析軟體等,增加了分析上的困難度。為了簡化及加速次世代定序資料的分析,目前已有許多自動化次世代定序資料分析的流程軟體,但這些軟體功能多注重在上游的序列分析如基因型的決定等,在決定基因型後,如何快速找到患病親屬間共有的基因突變,仍是分析次世代定序家族資料的挑戰。本院群體健康科學研究所鍾仁華副研究員及其團隊針對家族資料,開發了一套自動化次世代定序分析軟體,分析流程如圖一所示,此軟體結合了各種外部資料,例如自公用資料庫1000 Genomes Project所估算出的基因突變頻率、自SeattleSeq所取得的基因功能註解、自Rutgers Map所下載的基因位點等,依使用者的資料型態,若家族數不多,可執行染色體同源性分析(IBD analysis),找出患病親屬間高度共享的染色體片段,接著,可假設疾病模型(顯性或隱性)更進一步的找出致病位點。若家族數夠多,則可執行統計上的連鎖分析(linkage analysis)及關聯分析(association analysis),找出統計上顯著的突變位點。
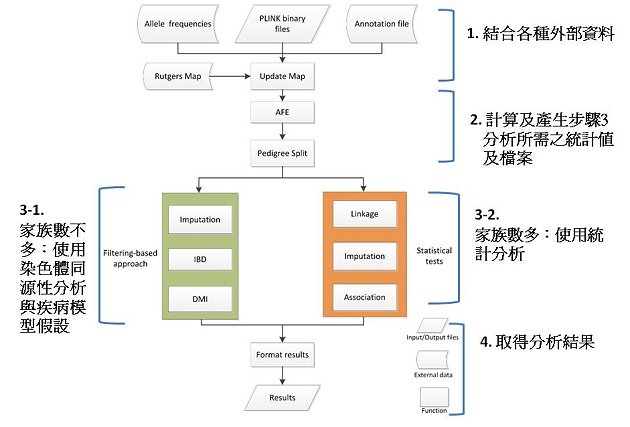
圖一:自動化次世代定序分析軟體分析流程圖
鍾博士團隊並與同所蔡慧如副研究員及長庚醫院精神科陳嘉祥醫師合作,將所發展的軟體應用在患有躁鬱症的家族上,透過染色體同源性分析、疾病模型假設及基因功能過濾後,找出在GBP5及DPP4基因中,有超過八成的患病親屬觀察到兩個罕見突變(突變頻率在一般族群裡小於0.5%),圖二紅框中顯示其中一個突變在該家族中的遺傳狀態。
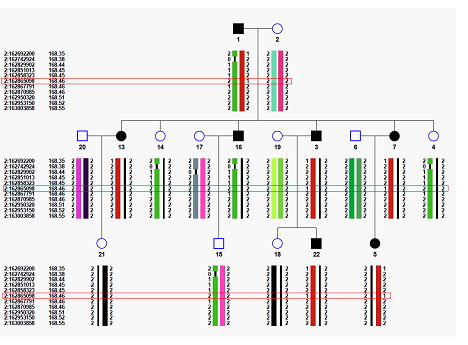
圖二:運用此軟體找出在第二條染色體上與躁鬱症相關的基因突變位點(紅框處)
此自動化次世代定序分析軟體已上線(http://fampipe.sourceforge.net)供研究人員免費下載,此研究論文已於今年6月發表於PLOS Computational Biology。
《文/圖:群體健康科學研究所鍾仁華副研究員》