NHRI Communications

知識產權
新藥再創新猷—漫談2015年美國FDA核准小分子新藥(下)
Introduction to FDA approved novel small molecular drugs in 2015 (part 3)
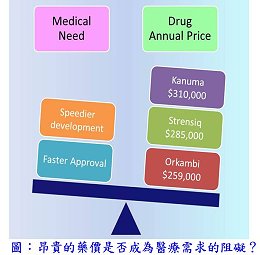 隨著審查效能的提升,藥物自研發到獲得核准的時程已有效縮短,對患者而言,實是一大福音;然而,高單價卻也引發另一個無法承擔之重,2015年獲核准之新藥中有12個藥物之藥價1年高於10萬美元[註1]。本期介紹的9個新藥當中有3個(Cholbam® 、Orkambi® 和Xuriden® )是用於治療患者總數少於20萬人的罕見疾病藥物,當中的Xuriden® 更是被用於超級罕見疾病(ultra-rare disease)—遺傳性乳清酸尿症(hereditary orotic aciduria)的治療,全世界僅有約二十名患者,而Orkambi® 則是2015年獲核准新藥中的第三高藥價,1年需25.9萬美元。
隨著審查效能的提升,藥物自研發到獲得核准的時程已有效縮短,對患者而言,實是一大福音;然而,高單價卻也引發另一個無法承擔之重,2015年獲核准之新藥中有12個藥物之藥價1年高於10萬美元[註1]。本期介紹的9個新藥當中有3個(Cholbam® 、Orkambi® 和Xuriden® )是用於治療患者總數少於20萬人的罕見疾病藥物,當中的Xuriden® 更是被用於超級罕見疾病(ultra-rare disease)—遺傳性乳清酸尿症(hereditary orotic aciduria)的治療,全世界僅有約二十名患者,而Orkambi® 則是2015年獲核准新藥中的第三高藥價,1年需25.9萬美元。(1) Kybella® (deoxycholic acid)
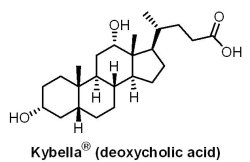 2015年4月29日,美國FDA 核准第一個以注射方式治療消除雙下巴的藥物Kybella® (deoxycholic acid, ATX-101),適用於下巴囤積中度或重度脂肪的成人。Kybella® 係屬合成的脫氧膽酸,也是人體自然產生的化合物,有助於人體吸收脂肪。此藥藥理作用藉由分解細胞膜,破壞脂肪細胞壁,從而溶解脂肪細胞,改善雙下巴。患者於單次治療中最多可能需注射50次(10 mL),在施打間隔不少於1個月的條件下,最多可進行6次單次治療。Kybella® 是由美國加州Kythera生技醫藥公司研發,但在2010年8月31日,以3.73億美元(含0.43億技轉金與3.3億里程碑金)和兩位數銷售權利金,將Kybella® (除美國與加拿大的權利)技轉給Bayer公司旗下負責皮膚業務Intendis公司。然而,有趣的是,Kythera公司在2014年3月10日又從Bayer公司手上將Kybella® 權利全部買回,Bayer公司獲得等值的0.33億美元Kythera股票、價值0.51億美元的本票(支付期限到2024年)和可能的銷售里程碑金。市場預估Kybella® 1年銷售額為3億美元。
2015年4月29日,美國FDA 核准第一個以注射方式治療消除雙下巴的藥物Kybella® (deoxycholic acid, ATX-101),適用於下巴囤積中度或重度脂肪的成人。Kybella® 係屬合成的脫氧膽酸,也是人體自然產生的化合物,有助於人體吸收脂肪。此藥藥理作用藉由分解細胞膜,破壞脂肪細胞壁,從而溶解脂肪細胞,改善雙下巴。患者於單次治療中最多可能需注射50次(10 mL),在施打間隔不少於1個月的條件下,最多可進行6次單次治療。Kybella® 是由美國加州Kythera生技醫藥公司研發,但在2010年8月31日,以3.73億美元(含0.43億技轉金與3.3億里程碑金)和兩位數銷售權利金,將Kybella® (除美國與加拿大的權利)技轉給Bayer公司旗下負責皮膚業務Intendis公司。然而,有趣的是,Kythera公司在2014年3月10日又從Bayer公司手上將Kybella® 權利全部買回,Bayer公司獲得等值的0.33億美元Kythera股票、價值0.51億美元的本票(支付期限到2024年)和可能的銷售里程碑金。市場預估Kybella® 1年銷售額為3億美元。(2) Cholbam® (cholic acid)
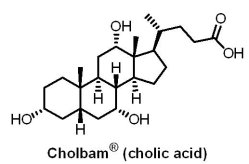 2015年3月17日,美國FDA 核准總部位於美國巴爾的摩的Asklepion製藥公司所研發用於治療罕見疾病—膽酸合成障礙(bile acid synthesis disorders)的市場首見(first-in-class)藥物Cholbam®(cholic acid)上市。Cholbam® 作為一種口服膠囊藥物,有效成分cholic acid本身是一個已知的膽酸。Cholbam® 用於治療因單一酶缺乏而導致膽酸合成障礙的兒科與成年患者及用於過氧化物酶體病(peroxisomal disorders),包括齊薇格譜系障礙(Zellweger spectrum disorders)。患有這類罕見疾病的患者因體內缺乏用來合成膽酸的酶,使得膽酸缺乏導致膽汁流量減少,從而致使具潛在毒性的膽酸中間體積聚於肝臟內(膽汁阻塞),並且對飲食中的脂肪和脂溶性維生素有吸收不良的問題。如果不進行治療,患者有肝臟疾病、脂肪痢(糞便中有脂肪)及脂溶性維生素吸收降低導致的併發症。Cholbam® 用於50名有18年以上治療史,因單一酶缺乏而導致膽酸合成障礙患者,其中有64%的患者出現反應,三分之二患者的存活期超過三年;另Cholbam® 用於29位有18年以上治療史,有過氧化物酶體病(包括齊薇格譜系障礙)的患者中,有46%的患者出現反應,42%患者的存活期超過三年。
2015年3月17日,美國FDA 核准總部位於美國巴爾的摩的Asklepion製藥公司所研發用於治療罕見疾病—膽酸合成障礙(bile acid synthesis disorders)的市場首見(first-in-class)藥物Cholbam®(cholic acid)上市。Cholbam® 作為一種口服膠囊藥物,有效成分cholic acid本身是一個已知的膽酸。Cholbam® 用於治療因單一酶缺乏而導致膽酸合成障礙的兒科與成年患者及用於過氧化物酶體病(peroxisomal disorders),包括齊薇格譜系障礙(Zellweger spectrum disorders)。患有這類罕見疾病的患者因體內缺乏用來合成膽酸的酶,使得膽酸缺乏導致膽汁流量減少,從而致使具潛在毒性的膽酸中間體積聚於肝臟內(膽汁阻塞),並且對飲食中的脂肪和脂溶性維生素有吸收不良的問題。如果不進行治療,患者有肝臟疾病、脂肪痢(糞便中有脂肪)及脂溶性維生素吸收降低導致的併發症。Cholbam® 用於50名有18年以上治療史,因單一酶缺乏而導致膽酸合成障礙患者,其中有64%的患者出現反應,三分之二患者的存活期超過三年;另Cholbam® 用於29位有18年以上治療史,有過氧化物酶體病(包括齊薇格譜系障礙)的患者中,有46%的患者出現反應,42%患者的存活期超過三年。成立於2006年的Asklepion是一家小型生技公司,主要研發領域為兒童罕見疾病。在Cholbam® 獲U.S. FDA核准前,Retrophin藥廠已於2015年1月12日自Asklepion公司取得cholic acid的專屬採購授權(exclusive right to purchase)。然而Retrophin藥廠過去是一個很有爭議性的公司,是對沖基金(Hedge Fund)爭議人物Martin Shkreli在2011年所創辦,2014年買下罕見疾病胱氨酸尿症(cystinuria)專藥Thiola® (tiopronin)之後,將藥價提高為原先20倍,從每劑1.50美元變成30美元。Shkreli又對外宣稱依據藥業的潛規則,藥廠會跟保險公司、醫院私下「喬」好價格,患者不用擔心會多付錢。此話一出,Shkreli簡直成了業界公敵,立刻遭董事會開除。之後Shkreli自創Turing 製藥公司,不改將患者當商品的惡性,重施故技,2015年8月買下上市已62年的Daraprim®(pyrimethamine)獨家行銷權,並於不久後將價格從每錠13.50美元提到750美元,漲幅將近55倍。此舉讓他下台一鞠躬,並再度成為輿論轟炸的對象,甚至還引起美國民主黨總統候選人希拉蕊批評,表示將提出抑制處方藥價格之方案。
當Cholbam® 被U.S. FDA以兒童罕見病核准之後,Retrophin藥廠亦透過Asklepion公司取得優先審查憑證(priority review voucher, PRV;為一種激勵措施,授予在熱帶病和罕見兒科疾病領域進行研究的單位,它可以使藥物的審查時間縮短至6個月)。在2015年5月27日Retrophin藥廠成功地以2.45億美元將此PRV售予法國藥廠巨擘Sanofi藥廠,Sanofi藥廠將自現階段13個後期產品線中,選擇最需要的產品透過此激勵憑證進行審查加持。其實這已不是Sanofi藥廠第一次通過購買憑證以加速其產品開發,在2014年7月也曾支付6.75億美元向BioMarin購買憑證,加快其降膽固醇藥Praluent® (alirocumab)審查。由於縮短4個月的審查期,最終領先競爭對手Amgen公司的降膽固醇藥Repatha® (evolocumab),提前1個月獲U.S. FDA核准上市。此外,在2014年11月19日加拿大蒙特婁Knight Therapeutics公司以1.25億美元出售因開發治療利什曼原蟲症的藥物Impavido® 而獲得的PRV予Gilead Sciences公司。而2015年3月10日United Therapeutics公司因開發用於兒童神經細胞瘤的藥物Unituxin® (dinutuximab)所獲得的一張兒科 PRV,目前尚待價而沽。
(3) ViberziTM (eluxadoline)
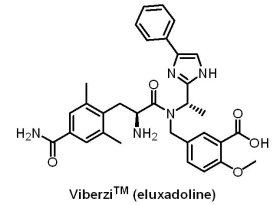 2015年5月27日,U.S. FDA核准Furiex製藥公司發展的ViberziTM(eluxadoline, JNJ-27018966)上市,用於治療腹瀉型腸激躁症(irritable bowel syndrome with diarrhea, IBS-D)。IBS-D是一種多因素腸胃道紊亂疾病,導致持續性腹痛、腹瀉並影響正常的腸道功能,女性病例數約為男性的2倍,在美國,約10%~15%的成人患有IBS-D,目前美國患者總數達1,500萬,歐洲患者總數達1,300萬。ViberziTM是一種(first-in-class)的口服藥物,每次服用2次,是μ和κ鴉片受體活化劑(μ , κ-opioid receptor agonist)與δ鴉片受體拮抗劑(δ-opioid receptor antagonist)。在臨床III期試驗中,有2,425例IBS腹瀉患者接受了1到2個療程(一個療程為26週)的ViberziTM或安慰劑,結果顯示ViberziTM對約30%的患者有效,有效率只比安慰劑高10%,因此,實際有效率只有10%。
2015年5月27日,U.S. FDA核准Furiex製藥公司發展的ViberziTM(eluxadoline, JNJ-27018966)上市,用於治療腹瀉型腸激躁症(irritable bowel syndrome with diarrhea, IBS-D)。IBS-D是一種多因素腸胃道紊亂疾病,導致持續性腹痛、腹瀉並影響正常的腸道功能,女性病例數約為男性的2倍,在美國,約10%~15%的成人患有IBS-D,目前美國患者總數達1,500萬,歐洲患者總數達1,300萬。ViberziTM是一種(first-in-class)的口服藥物,每次服用2次,是μ和κ鴉片受體活化劑(μ , κ-opioid receptor agonist)與δ鴉片受體拮抗劑(δ-opioid receptor antagonist)。在臨床III期試驗中,有2,425例IBS腹瀉患者接受了1到2個療程(一個療程為26週)的ViberziTM或安慰劑,結果顯示ViberziTM對約30%的患者有效,有效率只比安慰劑高10%,因此,實際有效率只有10%。值得一提的是,ViberziTM最早是由Johnson & Johnson子公司Janssen製藥公司開發,在2011年Janssen製藥公司決定聚焦發展抗病毒藥物與疫苗,於是將ViberziTM所有權轉給位於美國北卡的Furiex製藥公司。當Actavis藥廠在2014年7月1日耗資280億美元收購Forest Laboratories公司,隔天旋即耗資11億美元現金以及0.36億美元有價值權利(contingent value right,簡稱CVR)收購Furiex製藥公司;之後,Actavis藥廠在2015年3月17日以705億美元併購Allergan公司,並改名為Allergan,故ViberziTM的所有權經過多次易手,現由Allergan公司收入囊中,預計銷售額高峰值為4.5億美元。
(4) Orkambi® (lumacaftor/ivacaftor)
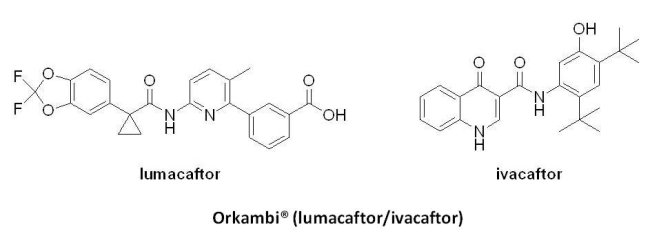
Vertex公司的Orkambi® (lumacaftor 200毫克/ivacaftor 125毫克)於2015年7月2日獲U.S. FDA核准成為首個用於治療12歲以上、囊腫纖化跨膜傳導調節蛋白(cystic fibrosis transmembrane conductance regulator, CFTR)基因存在F508del突變的囊狀纖維化症(cystic fibrosis, CF)患者之藥物。囊狀纖維化症是由於患者的第7對染色體長臂上的囊腫纖化跨膜傳導調節蛋白基因缺陷所造成的罕見遺傳疾病,全球約有七萬至十萬個病例,其中美國約有三萬名,具F508del雙基因突變者約占50%。Orkambi® 是Vertex公司結合已上市產品 Kalydeco® (ivacaftor)和lumacaftor所發展而成的複方藥物,此藥物獲得U.S. FDA的突破性藥物與孤兒藥資格,並通過優先審查程序。Orkambi® 在1,108位12歲以上、具F508del突變的囊狀纖維化症受試者參與的臨床試驗中被證實能改善患者的肺功能,常見的副作用為呼吸急促、上呼吸道感染、噁心、腹瀉、皮疹及女性月經異常。然而,於2016年,英國國家健康暨臨床醫學研究院(National Institute for Health and Clinical Excellence, NICE)以Orkambi® 雖能降低囊狀纖維化症患者因病情惡化而住院的頻率,於肺功能的改善卻很有限且價格過高為由,拒絕了此藥物的申請。
(5) AddyiTM (flibanserin)
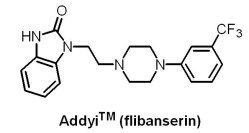 U.S. FDA於2015年8月18日核准Sprout製藥公司的AddyiTM(flibanserin)用於治療停經前女性的性慾低落障礙(hypoactive sexual desire disorder, HSDD)。AddyiTM原是Boehringer Ingelheim公司研發用於抗抑鬱的藥物,雖效果不彰,卻意外發現具有提高女性性慾的效果,暱稱為「女性威而鋼」。AddyiTM為一個突觸後5-HT1A受體活化劑與5-HT2A受體拮抗劑,然其改善性慾與相關困擾之機制仍未清楚。此外,肝功能不全之患者或在使用AddyiTM治療期間同時服用酒精或CYP3A4抑制劑之患者,可能有嚴重低血壓與昏厥之風險,已於仿單之加框警語(boxed warning)詳列可能之發生風險。此外,U.S. FDA要求Sprout製藥公司進行3項具良好設計、針對女性之臨床試驗,以進一步瞭解AddyiTM與酒精相互作用產生之已知嚴重風險。由於安全性的考量,U.S. FDA曾於2010年首度拒絕了AddyiTM的申請,隨後接手AddyiTM 的Sprout 製藥公司於2013年的申請再次被U.S. FDA拒絕,最後在2015年以醫師開立處方時應告知患者藥物副作用風險的前提下,U.S. FDA核准了該藥的上市申請。
U.S. FDA於2015年8月18日核准Sprout製藥公司的AddyiTM(flibanserin)用於治療停經前女性的性慾低落障礙(hypoactive sexual desire disorder, HSDD)。AddyiTM原是Boehringer Ingelheim公司研發用於抗抑鬱的藥物,雖效果不彰,卻意外發現具有提高女性性慾的效果,暱稱為「女性威而鋼」。AddyiTM為一個突觸後5-HT1A受體活化劑與5-HT2A受體拮抗劑,然其改善性慾與相關困擾之機制仍未清楚。此外,肝功能不全之患者或在使用AddyiTM治療期間同時服用酒精或CYP3A4抑制劑之患者,可能有嚴重低血壓與昏厥之風險,已於仿單之加框警語(boxed warning)詳列可能之發生風險。此外,U.S. FDA要求Sprout製藥公司進行3項具良好設計、針對女性之臨床試驗,以進一步瞭解AddyiTM與酒精相互作用產生之已知嚴重風險。由於安全性的考量,U.S. FDA曾於2010年首度拒絕了AddyiTM的申請,隨後接手AddyiTM 的Sprout 製藥公司於2013年的申請再次被U.S. FDA拒絕,最後在2015年以醫師開立處方時應告知患者藥物副作用風險的前提下,U.S. FDA核准了該藥的上市申請。(6) Xuriden® (uridine triacetate)
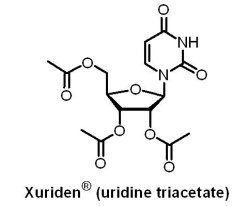 Wellstat Therapeutics公司的Xuriden® (uridine triacetate)於2015年9月4日通過U.S. FDA優先審查途徑,用於超級罕見疾病—遺傳性乳清酸尿症的治療,是第一個獲准用於此疾病的藥物;Wellstat Therapeutics公司也因此取得一張罕見兒科疾病優先審查憑證。遺傳性乳清酸尿症是一種罕見的遺傳代謝疾病,全世界僅有約二十例患者,因合成核糖核酸(RNA)之必要成分尿苷(uridine)的合成酶有缺陷或短少所致。其臨床症狀為貧血、白血球數減少、嗜中性白血球數減少、乳清酸晶體形成造成尿道阻塞與發育遲緩或停滯;臨床治療方式為補充尿苷,以促進體內乳清酸轉變為尿苷酸(uridylic acid),進而降低尿液中的乳清酸,減緩相關症狀。Xuriden® 為一個口服尿苷三乙酸酯(uridine triacetate),可於人體內代謝為尿苷,具有優於尿苷的吸收效果。自僅4名患者(3至19歲)的臨床試驗結果顯示,使用Xuriden® 的受試者均可表現穩定的血液學參數,且未觀察到不良反應。
Wellstat Therapeutics公司的Xuriden® (uridine triacetate)於2015年9月4日通過U.S. FDA優先審查途徑,用於超級罕見疾病—遺傳性乳清酸尿症的治療,是第一個獲准用於此疾病的藥物;Wellstat Therapeutics公司也因此取得一張罕見兒科疾病優先審查憑證。遺傳性乳清酸尿症是一種罕見的遺傳代謝疾病,全世界僅有約二十例患者,因合成核糖核酸(RNA)之必要成分尿苷(uridine)的合成酶有缺陷或短少所致。其臨床症狀為貧血、白血球數減少、嗜中性白血球數減少、乳清酸晶體形成造成尿道阻塞與發育遲緩或停滯;臨床治療方式為補充尿苷,以促進體內乳清酸轉變為尿苷酸(uridylic acid),進而降低尿液中的乳清酸,減緩相關症狀。Xuriden® 為一個口服尿苷三乙酸酯(uridine triacetate),可於人體內代謝為尿苷,具有優於尿苷的吸收效果。自僅4名患者(3至19歲)的臨床試驗結果顯示,使用Xuriden® 的受試者均可表現穩定的血液學參數,且未觀察到不良反應。(7) Veltassa® (patiromer)
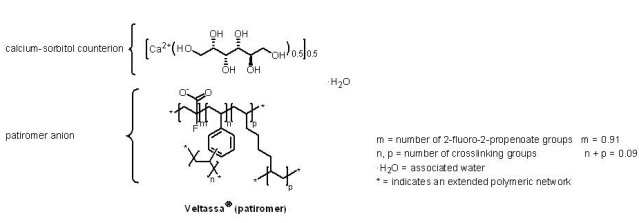
2015年10月21日Relypsa公司的Veltassa® (patiromer)獲U.S. FDA核准用於高血鉀症(hyperkalemia)之治療,由於鉀離子可以調節細胞內之滲透壓與人體酸鹼值的平衡,亦具有幫助神經衝動傳導與維持肌肉與心肌的功能,同時亦為細胞生長及代謝所必需的元素,當血清中的鉀離子濃度超過5.5 mEq/L(正常為3.5-5.5 mEq/L)即可診斷為高血鉀。腎臟對鉀離子具有調控作用,體內的鉀離子有80~90%是由腎臟經尿液排出,當腎臟功能低下時,便可能導致血液中鉀離子濃度過高;此疾病最容易發生在急性或慢性腎臟病與心臟病患者身上,尤其是有服用可調節血壓和體液平衡(fluid balance)的腎素—血管收縮素—醛固酮系統抑制劑(renin-angiotensin-aldosterone system inhibitor, RAAS inhibitor)的患者。Veltassa® 為一個非吸收、含鈣—山梨醇的陽離子交換聚合物(non-absorbed, cation exchange polymer that contains a calcium-sorbitol),是粉末狀藥物,須與水混合後服用,藉由在胃腸道與鉀離子結合,以減少鉀離子的吸收。臨床試驗結果顯示,Veltassa® 可降低使用至少一種腎素—血管收縮素—醛固酮系統抑制劑的慢性腎臟病患者血清中之鉀含量,而最常見的不良反應為便秘、低鎂血症(hypomagnesemia)、腹瀉、噁心、腹部不適與脹氣。由於Veltassa® 的藥效起始較慢,故不建議作為高血鉀症的緊急用藥;此外,審查過程中發現Veltassa® 與許多藥物都有交互作用,因而降低其藥效,所以U.S. FDA建議Veltassa® 的服用至少須與其他口服藥間隔6小時以上,並已以黑框警告。
(8) Zurampic® (lesinurad)
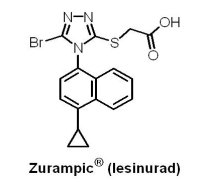 2015年12月22日U.S. FDA 核准AstraZeneca公司的Zurampic® (lesinurad)與可減少尿酸產生的黃嘌呤氧化酶抑制劑(xanthine oxidase inhibitor, XOI)合併用於痛風相關高尿酸血症(hyperuricemia)的治療。當體內尿酸累積於血液中無法隨尿液排出時,即稱為高尿酸血症,多數高尿酸血症並不會發展成痛風,只有當形成尿酸鹽結晶沉積在關節腔內時,才可能造成痛風。Zurampic® (lesinurad)是一種選擇性尿酸再吸收抑制劑(selective uric acid reabsorption inhibitor, SURI),藉由抑制尿酸鹽轉運蛋白1(urate transporter 1, URAT1)再吸收尿酸的功能,促進尿酸排泄,進而降低血中尿酸濃度。在1,537位受試者、長達12個月的3個臨床試驗中,Zurampic® 與黃嘌呤氧化酶抑制劑合併使用被證實具有降低血清中尿酸含量的效果,常見的不良反應為頭痛、流行性感冒、血清肌酸酐(creatinine)增加以及胃食道逆流。Zurampic® 的黑框警告內容指出,當未合併使用黃嘌呤氧化酶抑制劑且給予較核准劑量高的Zurampic® 時,會增加急性腎功能衰竭的風險;此外,U.S. FDA要求AstraZeneca公司須進行Zurampic® 上市後,服用此藥物患者的腎臟和心血管安全性評估。
2015年12月22日U.S. FDA 核准AstraZeneca公司的Zurampic® (lesinurad)與可減少尿酸產生的黃嘌呤氧化酶抑制劑(xanthine oxidase inhibitor, XOI)合併用於痛風相關高尿酸血症(hyperuricemia)的治療。當體內尿酸累積於血液中無法隨尿液排出時,即稱為高尿酸血症,多數高尿酸血症並不會發展成痛風,只有當形成尿酸鹽結晶沉積在關節腔內時,才可能造成痛風。Zurampic® (lesinurad)是一種選擇性尿酸再吸收抑制劑(selective uric acid reabsorption inhibitor, SURI),藉由抑制尿酸鹽轉運蛋白1(urate transporter 1, URAT1)再吸收尿酸的功能,促進尿酸排泄,進而降低血中尿酸濃度。在1,537位受試者、長達12個月的3個臨床試驗中,Zurampic® 與黃嘌呤氧化酶抑制劑合併使用被證實具有降低血清中尿酸含量的效果,常見的不良反應為頭痛、流行性感冒、血清肌酸酐(creatinine)增加以及胃食道逆流。Zurampic® 的黑框警告內容指出,當未合併使用黃嘌呤氧化酶抑制劑且給予較核准劑量高的Zurampic® 時,會增加急性腎功能衰竭的風險;此外,U.S. FDA要求AstraZeneca公司須進行Zurampic® 上市後,服用此藥物患者的腎臟和心血管安全性評估。(9) Bridion® (sugammadex)
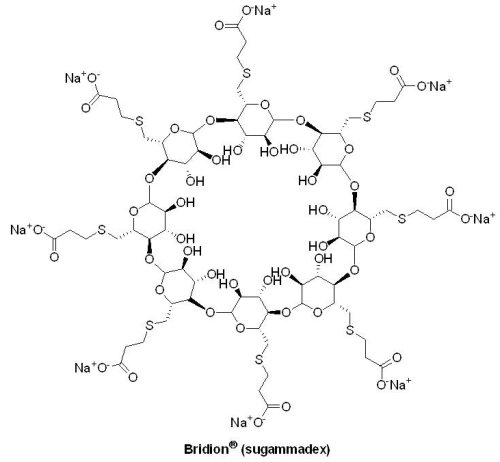 Merck藥廠的Bridion® (sugammadex)於2015年12月15日獲U.S. FDA 核准用於成人於特定手術期間因肌肉鬆弛劑「羅庫溴銨(rocuronium bromide)或維庫溴銨(vecuronium bromide)」誘導的深度神經肌肉阻斷(neuromuscular blockade)之逆轉藥物;於此之前,Bridion® 已於全球五十多個國家獲准上市,然而基於安全性的考量,U.S. FDA曾分別於2008與2013年拒絕此藥物的上市申請。Bridion® 最初是Organon藥廠所開發的藥物,Organon藥廠於2007年被Schering-Plough藥廠收購,隨後Merck藥廠於2009年併購Schering-Plough藥廠,因而取得Bridion® 的所有權。Bridion® 為選擇性肌肉鬆弛結合劑γ-cyclodextrin的類似物,藉由在血漿中與羅庫溴銨或維庫溴銨形成複合物,降低血漿中肌肉鬆弛劑的濃度,進而逆轉其藥效。在總共456位受試者的3個臨床III期試驗結果顯示,大部分受試者在常規使用 Bridion® 5 分鐘內即能恢復,常見的不良反應為嘔吐、低血壓、疼痛、頭痛以及噁心。在一個評估嚴重、可能危及生命的過敏反應(anaphylaxis)與超敏反應(hypersensitivity reactions)的臨床試驗結果顯示,299名受試者中,有1名使用 Bridion® 的受試者出現嚴重的過敏反應,因此提醒臨床醫師應警覺過敏反應發生的可能性並採取適當的措施。此外,在使用Bridion® 數分鐘之後,有患者出現心博徐緩(bradycardia)的問題,其中更有出現心跳停止(cardiac arrest)的例子,因此建議醫師在觀察到相關問題時,須密切監控患者的血液動力學變化(hemodynamic changes),並給予抗膽鹼藥物(anticholinergic agents)。因Bridion® 可能會暫時性降低激素類避孕藥物的效果,所以建議使用該類避孕藥的女性在使用Bridion® 期間改以另一種避孕方法。
Merck藥廠的Bridion® (sugammadex)於2015年12月15日獲U.S. FDA 核准用於成人於特定手術期間因肌肉鬆弛劑「羅庫溴銨(rocuronium bromide)或維庫溴銨(vecuronium bromide)」誘導的深度神經肌肉阻斷(neuromuscular blockade)之逆轉藥物;於此之前,Bridion® 已於全球五十多個國家獲准上市,然而基於安全性的考量,U.S. FDA曾分別於2008與2013年拒絕此藥物的上市申請。Bridion® 最初是Organon藥廠所開發的藥物,Organon藥廠於2007年被Schering-Plough藥廠收購,隨後Merck藥廠於2009年併購Schering-Plough藥廠,因而取得Bridion® 的所有權。Bridion® 為選擇性肌肉鬆弛結合劑γ-cyclodextrin的類似物,藉由在血漿中與羅庫溴銨或維庫溴銨形成複合物,降低血漿中肌肉鬆弛劑的濃度,進而逆轉其藥效。在總共456位受試者的3個臨床III期試驗結果顯示,大部分受試者在常規使用 Bridion® 5 分鐘內即能恢復,常見的不良反應為嘔吐、低血壓、疼痛、頭痛以及噁心。在一個評估嚴重、可能危及生命的過敏反應(anaphylaxis)與超敏反應(hypersensitivity reactions)的臨床試驗結果顯示,299名受試者中,有1名使用 Bridion® 的受試者出現嚴重的過敏反應,因此提醒臨床醫師應警覺過敏反應發生的可能性並採取適當的措施。此外,在使用Bridion® 數分鐘之後,有患者出現心博徐緩(bradycardia)的問題,其中更有出現心跳停止(cardiac arrest)的例子,因此建議醫師在觀察到相關問題時,須密切監控患者的血液動力學變化(hemodynamic changes),並給予抗膽鹼藥物(anticholinergic agents)。因Bridion® 可能會暫時性降低激素類避孕藥物的效果,所以建議使用該類避孕藥的女性在使用Bridion® 期間改以另一種避孕方法。新藥研發在生技醫藥產業連續兩年的大放異彩,不僅提升了相關產業的信心,對人民的健康福祉亦多了一分保障。除了罕見疾病藥物與醫療需求藥物依然是備受矚目的焦點,併購、技轉或共同合作開發仍是2015年獲核准新藥的重要一環,反映出新藥開發的耗時與高風險,而大藥廠的豐沛資源與臨床經驗對整個開發過程著實有其優勢;此外,不少藥物都有在臨床III期失敗的紀錄,但透過重新設計臨床III期試驗,找出該藥物之特異性,最終取得新藥上市的鑰匙。不管如何,在審查效能與臨床需求並進的同時,我們能預見新藥研發將持續蓬勃發展,在眾多政策的支持與鼓勵下,以往未受關注的疾病用藥或許不再遙遙無期,只是藥物價格是否能符合大眾的期待,恐怕是未來世界各國在醫藥給付所需要面對的難題。
註1:Jarvis, L. M. The Year in New Drugs. C&EN 2016;94:12-17.
《文/圖:生技與藥物研究所謝興邦研究員、李靜琪副研究員;審校:生技與藥物研究所陳炯東研究員》