NHRI Communications

知識產權
新藥回歸正常—漫談2016年FDA核准小分子新藥(上)
Introduction to FDA approved novel small molecular drugs in 2016 (part 1)
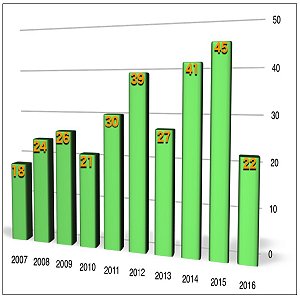 繼2014年與2015年新藥核准大爆發(分別核准41個新藥「註1」與45個新藥「註2」)後,2016年可說是從絢爛回歸正常,核准案件(22個)甚至比過去10年的平均案件(30個新藥卅年)還來得低。美國食品藥物管理局(Food and Drug Administration, FDA)新藥部門John Jenkins主任就此結果表示,審查截止日期原訂於2016年的5個藥物,在加速審查機制的優惠下,於2015年年底前獲准上市;此外,相較於2014年申請的41件與2015年申請的40件,2016年共有36件新藥申請案,案件並未有明顯落差。然而,因為2016年FDA共發出44封補件信函給申請新藥查驗登記(new drug application, NDA)的廠商,其中包括14封完整補件通知(complete response letters),相較於2015年的20封補件信函中,僅有2封完整補件通知來的多。在新藥審查退件數較高,且5個藥物提前於2015年核准通過的結果下,導致2016年整體通過案件數低於過去10年的平均件數。
繼2014年與2015年新藥核准大爆發(分別核准41個新藥「註1」與45個新藥「註2」)後,2016年可說是從絢爛回歸正常,核准案件(22個)甚至比過去10年的平均案件(30個新藥卅年)還來得低。美國食品藥物管理局(Food and Drug Administration, FDA)新藥部門John Jenkins主任就此結果表示,審查截止日期原訂於2016年的5個藥物,在加速審查機制的優惠下,於2015年年底前獲准上市;此外,相較於2014年申請的41件與2015年申請的40件,2016年共有36件新藥申請案,案件並未有明顯落差。然而,因為2016年FDA共發出44封補件信函給申請新藥查驗登記(new drug application, NDA)的廠商,其中包括14封完整補件通知(complete response letters),相較於2015年的20封補件信函中,僅有2封完整補件通知來的多。在新藥審查退件數較高,且5個藥物提前於2015年核准通過的結果下,導致2016年整體通過案件數低於過去10年的平均件數。2016年FDA所核准的22個新藥,包括7個生物藥和15個小分子藥物,依其特點可歸類如下:(a)治療癌症、感染症與神經疾病藥物數目居首,各占18%(4個);顯影劑與治療皮膚疾病藥物次之,各占9%(2個);(b) 8個新藥(36%)為具新機轉的市場首見(first-in-class)新藥(36%-2015年),包括以下藥物:Defitelio®、Exondys 51TM、Ocaliva®、Spinraza®、VenclextaTM、Xiidra®、Zinbryta®、ZinplavaTM;(c) 9個新藥(41%)是用於治療患者總數少於20萬人的罕見疾病用藥(47%-2015年),包括以下藥品:Anthim®、Defitelio®、 Exondys 51TM、LartruvoTM、NetspotTM、Ocaliva®、Rubraca®、Spinraza®、VenclextaTM。其中值得一提的是,Exondys 51TM—用於治療裘馨氏肌肉萎縮症(duchenne muscular dystrophy, DMD)患者、Spinraza®—用於治療脊髓性肌萎縮(spinal muscular atrophy, SMA)患者及Defitelio®—用於治療幹細胞移植造成的肝靜脈阻塞(hepatic veno-occlusive disease, VOD)患者,此3個藥物都是屬於寡核苷酸(oligonucleotide);(d) 16個新藥(73%)是獲得一種或一種以上加速方案:快速通道(fast track)、突破性療法(breakthrough)、優先審查(priority review)及加速核准(accelerated approval)資格的新藥(60%-2015年),分別有36%(8個)、32%(7個)、68%(15個)和27%(15個)。
2016年核准的15個小分子藥物,除上述3個寡核苷酸外,亦包括1個胜肽(peptide)新藥AdlyxinTM與2個用於PET之顯影劑,前者為用於治療第2型糖尿病的類升糖素胜肽-1受體促效劑(glucagon-like peptide-1 agonist, GLP-1 agonist);後者分別是鎵68 DOTA-TATE的NetspotTM和氟18 Fluciclovine的AxuminTM。15個小分子新藥將分上中下三期進行詳細報導。
抗癌藥物
(1) Rubraca® (Rucaparib)
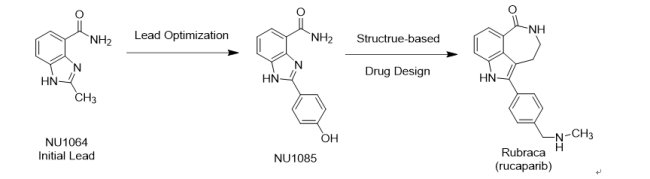
繼2014年12月19日英國AstraZeneca藥廠所開發的LynparzaTM(Olaparib;AZD-2281;KU 59436) 獲准成為第一個針對聚ADP-核糖聚合酶(poly ADP ribose polymerase, PARP)抑制劑上市後,FDA於2016年12月19日以突破性藥物資格、優先審查資格、孤兒藥地位和加速核准機制,核准由美國生技藥廠Clovis Oncology研發的第二個PARP抑制劑Rubraca® (Rucaparib;AG-014699;PF-01367338;CO-338)上市。口服錠劑Rubraca®用於治療曾接受兩種或更多化療方案、且帶有BRCA基因突變(約占所有卵巢癌患者的15 – 20%)的晚期卵巢癌患者。由於PARP抑制劑可以阻止參與修復受損DNA的酶活性,故通過阻斷這種酶時,帶有BRCA基因突變的癌細胞DNA較無法進行修復,最終導致癌細胞死亡、腫瘤生長減慢或停止生長。同時FDA也核准由Foundation Medicine開發的次世代定序(NGS的檢測方法)的伴隨式診斷試劑CDxBRCA,用於診斷帶有BRCA基因突變的晚期卵巢癌患者,只要受試者對CDxBRCA測試呈現陽性反應(證明其攜帶BRCA1 / 2基因突變),便適合接受Rubraca®的治療。由於LynparzaTM獲准用於治療曾接受三種以上化療方案的患者,而Rubraca®獲准用於治療曾接受二種以上化療方案的患者,故Rubraca®的上市,對於AstraZeneca藥廠的LynparzaTM將構成市場競爭威脅。目前AbbVie藥廠(Veliparib)、Pfizer藥廠(Talazoparib,併購Medivation取得)和Tesaro生技公司(Niraparib)都有PARP抑制劑在晚期臨床試驗階段,尤其最近Niraparib破天荒的臨床III期優異表現,使得PARP抑制劑的競爭也更加白熱化。
FDA核准Rubraca®是基於一項包括106位已接受二種或更多種化療但攜帶BRCA突變晚期卵巢癌患者的臨床III期研究結果。資料顯示,有54%接受Rubraca®治療的受試者腫瘤曾達到完全或部分縮小,反應持續中位數為9.2個月。在副作用方面值得注意的是,Rubraca®使用者會產生骨髓增生不良症候群(myelodysplastic syndrome, MDS)、急性骨髓性白血病(acute myeloid leukemia, AML)和胎兒傷害等嚴重風險。
Rubraca®的早期藥物開發是一個非常成功的產學合作案例,值得大家借鏡。在1993年左右,英國Newcastle University生物學Barbara Durkacz教授和Nicola Curtin教授及藥物化學Bernard Golding教授和Roger Griffin教授共同開發PARP抑制劑benzimidazole NU1064 和NU1085。在1997年10月2日,當時位於美國聖地牙哥La Jolla的製藥公司Agouron Pharmaceuticals與英國Newcastle University簽署共同合作開發PARP抑制劑合約書。由於Agouron公司專長為結構生物學之共結晶技術,因此很快就得到NU1085與PARP酵素共結晶,接著透過雙邊藥物化學的合作,設計新一代PARP抑制劑,進而順利開發出AG014699(Rubraca®)。而後續臨床前的動物藥理實驗以及臨床I期試驗,在英國癌症研究中心(Cancer Research UK)的經費支持下,由英國Newcastle University生物學Nicola Curtin教授和Herbie Newell教授及臨床學Ruth Plummer教授和Hilary Calvert教授領導執行,促使Rubraca®得以於2003年首次以靜脈注射並與口服temozolomide合併使用進入Phase 0/I試驗。
1999年1月27日,Warner-Lambert公司以21億美元收購Agouron Pharmaceuticals公司,一年後,Pfizer藥廠於2000年2月7日以900億美元併購Warner-Lambert公司,Rubraca®之所有權即轉由Pfizer藥廠擁有,並將代號更改成PF-01367338。至於Pfizer藥廠為何要將PF-01367338技轉給Clovis Oncology藥廠?這就得從Sanofi-Aventis公司的Iniparib(BSI-201)說起。話說,Sanofi-Aventis公司在2009年花費5億美元從BiPar Sciences公司取得PARP抑制劑 Iniparib,但是在2011年1月28日,Iniparib用於治療三陰性乳癌之臨床III期試驗解盲失敗,一時之間,大藥廠對於PARP抑制劑是否能闖過臨床試驗,產生很大的懷疑,以致Pfizer藥廠希望能將Rubraca®移出自家臨床產品線,隨即在2011年6月2日將Rubraca®技轉給Clovis Oncology藥廠,取得700萬簽約金,並簽訂3個階段性里程碑金(40萬、8,850萬和2,075萬美元)以及未來銷售權利金。當時Rubraca®口服製劑處於臨床I/II期,而靜脈製劑則在臨床II期階段,Clovis Oncology藥廠於是接手推動臨床試驗,除了推動Rubraca®在三陰性乳癌之應用,亦在2012年取得FDA授予孤兒藥地位用於卵巢癌之治療,最後終於在2016年年底順利搶下聖杯。目前,Rubraca®15天治療療程約需6,870美元,市場預估在2018年可達到2.67億美元年銷售額。
(2) VenclextaTM (Venetoclax)
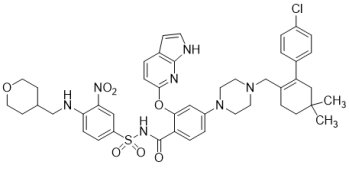 FDA於2016年4月11日以突破性藥物資格、優先審查資格、孤兒藥地位和加速核准機制,核准由AbbVie藥廠和Roche藥廠旗下Genentech合作開發的市場首見新藥VenclextaTM(Venetoclax;ABT-199),用於治療攜帶17p刪除突變(17p deletion)以及之前已接受至少一種療法的慢性淋巴細胞白血病(chronic lymphocytic leukemia, CLL)患者;同時,FDA亦核准由Abbott Molecular公司生產的伴隨診斷Vysis CLL FISH探針試劑盒,以確認攜帶17p刪除突變的CLL患者。VenclextaTM是B細胞淋巴瘤因子-2(B-Cell Lymphoma 2, BCL-2)抑制劑,亦是第一個獲核准上市的蛋白相互作用(Protein-Protein Interaction, PPI)抑制劑。由於BCL-2是一種抑制細胞凋亡蛋白,在某些癌細胞中(例如CLL)高度表達,並造成癌細胞產生相關抗藥性,故VenclextaTM利用選擇性抑制BCL-2的功能,幫助恢復讓白血病細胞進行自然凋亡的過程,達到治療腫瘤的目的。
FDA於2016年4月11日以突破性藥物資格、優先審查資格、孤兒藥地位和加速核准機制,核准由AbbVie藥廠和Roche藥廠旗下Genentech合作開發的市場首見新藥VenclextaTM(Venetoclax;ABT-199),用於治療攜帶17p刪除突變(17p deletion)以及之前已接受至少一種療法的慢性淋巴細胞白血病(chronic lymphocytic leukemia, CLL)患者;同時,FDA亦核准由Abbott Molecular公司生產的伴隨診斷Vysis CLL FISH探針試劑盒,以確認攜帶17p刪除突變的CLL患者。VenclextaTM是B細胞淋巴瘤因子-2(B-Cell Lymphoma 2, BCL-2)抑制劑,亦是第一個獲核准上市的蛋白相互作用(Protein-Protein Interaction, PPI)抑制劑。由於BCL-2是一種抑制細胞凋亡蛋白,在某些癌細胞中(例如CLL)高度表達,並造成癌細胞產生相關抗藥性,故VenclextaTM利用選擇性抑制BCL-2的功能,幫助恢復讓白血病細胞進行自然凋亡的過程,達到治療腫瘤的目的。美國國家癌症研究所(NCI)指出,CLL是成人中最常見的白血病類型,每年新確診的個案達15,000例。據估計,大約10%的初治(未經治療)CLL患者與大約20%的復發性CLL患者攜帶17p刪除突變,患者平均存活期通常不到3年。染色體17p中有著名的腫瘤抑制基因p53,其存在能大幅提高細胞對藥物的敏感度,因此,17p缺失的CLL患者對化療可能會不那麼敏感,容易產生抗藥性或者復發,故被認為預後最差的群組。
FDA是基於一項開放標籤、單組、多中心II期臨床研究(M13-982)數據核准VenclextaTM上市。在一項納入106位17p刪除突變並至少接受過一次其它治療的CLL患者的單藥臨床試驗中,患者每天口服VenclextaTM,初始劑量20 mg,經5週後增至400 mg,結果顯示,80%患者完全緩解或部分緩解,包括近20%完全緩解,2年存活率高達84%,而且副作用也較輕微。VenclextaTM能有這樣的結果非常驚人,因受試者都是其它療法失敗的晚期患者,故對很多患者而言,這可說是起死回生的仙丹。目前VenclextaTM 亦針對非何杰金氏淋巴瘤(non-hodgkin’s lymphoma, NHL)、瀰漫性大B細胞淋巴瘤(diffuse large b cell lymphoma, DLBCL)、急性骨髓性白血病(acute myeloid leukemia, AML)和多發性骨髓瘤(multiple myeloma, MM)進行臨床試驗,預估未來幾年將會取得其他癌症之適應症。目前VenclextaTM一年藥價為11萬美元,市場預估到2018年銷售額可達到14.8億美元。
AbbVie藥廠在2015年3月5日以破天荒210億美元買下Pharmacyclics生技公司,一方面是由於治療類風濕性關節炎暢銷藥Humira®(adalimumab)專利將於2017年到期,將面臨其他公司推出adalimumab生物類似藥的挑戰,恐將大幅減少Humira®營收,因此想透過Imbruvica®彌補其營收減少,另一方面亦可一舉進入血液癌症領域,以強化其腫瘤治療市場之競爭;隨著VenclextaTM上市,將與Imbruvica®成為AbbVie藥廠在血液癌症臨床治療的一流組合治療。
放射性診斷劑
(1) AxuminTM
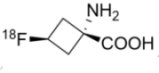 2016年5月27日,美國FDA核准由英國Blue Earth Diagnostics公司開發的AxuminTM (Fluciclovine (18F); anti-1-amino-3-18F-fluorocyclobutane-1-carboxylic acid (FACBC);anti-3[18F] FACBC or F18)為一種新型正子斷層掃描(positron emission tomography, PET)放射性診斷劑,藉由特定抗原(prostate-specific antigen, PSA)的含量檢測,評估前列腺癌復發風險。PSA之含量檢測是目前醫師們最常使用的篩檢方式,但由於PSA的數量會隨著年紀、疾病的進展而發生改變,使人們無法明確定位癌症細胞是否復發。據估計,前列腺癌患者在接受治療後,未来的10到15年内復發率接近三分之一,故在前列腺癌治療中,病灶的精確定位和發展階段的分析以及對於改善疾病的治療和結果,顯得尤為重要。
2016年5月27日,美國FDA核准由英國Blue Earth Diagnostics公司開發的AxuminTM (Fluciclovine (18F); anti-1-amino-3-18F-fluorocyclobutane-1-carboxylic acid (FACBC);anti-3[18F] FACBC or F18)為一種新型正子斷層掃描(positron emission tomography, PET)放射性診斷劑,藉由特定抗原(prostate-specific antigen, PSA)的含量檢測,評估前列腺癌復發風險。PSA之含量檢測是目前醫師們最常使用的篩檢方式,但由於PSA的數量會隨著年紀、疾病的進展而發生改變,使人們無法明確定位癌症細胞是否復發。據估計,前列腺癌患者在接受治療後,未来的10到15年内復發率接近三分之一,故在前列腺癌治療中,病灶的精確定位和發展階段的分析以及對於改善疾病的治療和結果,顯得尤為重要。一般PET腫瘤掃描診斷是使用18F-FDG為診斷劑,然而由於前列腺癌不消耗過多葡萄糖,故PET的定位效果不佳,不利於前列腺癌之影像診斷。C11 choline是2012年FDA核准用於前列腺癌可能復發之診斷劑,但由於半衰期僅20分鐘,故大大限制它在臨床的廣泛運用。AxuminTM的活性成分為fluciclovine F18,是一種非天然胺基酸,可被位於前列腺癌細胞表面的胺基酸轉運體吸收至癌細胞內。研究發現,前列腺癌細胞比其周圍的健康組織更易吸收fluciclovine F18,且在吸收後不會將其代謝或用於蛋白質合成。FDA的核准是基於AxuminTM作為診斷劑之兩項臨床實驗:實驗一為針對105位前列腺癌可能復發的患者進行掃描診斷,並和組織病理切片結果進行比較;實驗二為2種PET診斷劑(AxuminTM和已核准的C11 choline)影像診斷結果的比較。這兩項研究結果支持AxuminTM用於在先前治療後PSA水平升高之前列腺癌患者於病灶影像診斷的安全性與有效性。
AxuminTM這個小分子是由任職於美國Emory University的放射藥物化學家Mark Goodman博士與同事Tim Shoup博士設計開發,並於1996年提出專利申請,之後就技轉給長年與Goodman博士有產學合作的公司Nihon Medi-physics,2008年GE Health再從Nihon Medi-physics取得AxuminTM,並進行各種癌症影像診斷之臨床試驗,但由於所呈現之臨床效果並沒有比18F-FDG 好,再加上GE Health當時主要想發展阿茲海默疾病之影像診斷劑,故對於AxuminTM之臨床發展漸漸失去興趣。Mark Goodman 博士之後與同校影像暨放射科學副教授David Schuster醫師合作,發現AxuminTM在前列腺癌的影像診斷比18F-FDG來得更精準,於是在2014年時,當時任職於GE Health 分子影像PET小組負責人Jonathan Allis博士決定籌錢與一些GE員工成立新公司,將AxuminTM推向前列腺癌影像診斷領域。最後,由英國創投Syncona Partners資助2,150萬美元成立Blue Earth Diagnostics新創公司。值得一提的是,成立於2012年的Syncona Partners創投是屬於比爾蓋茲基金會所投資的Wellcome Trust。AxuminTM的成功上市,再為Emory University新增一筆上市藥物原始貢獻學校紀錄;2011年New England Journal of Medicine調查,過去40年共有153項 FDA核准的藥物與疫苗,而Emory University在其原始開發為大學或政府機構項目中排名第四(美國國家衛生研究院(NIH)有22項、The University of California System有11項、Memorial Sloan-Kettering有8項、Emory University有7項,Yale University有6項)。
(2) NetspotTM
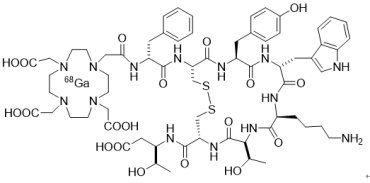 2016年6月1日,美國FDA以孤兒藥地位和優先審查資格核准由總部位於法國Advanced Accelerator Application USA公司開發的一款新的診斷顯像劑NetspotTM(gallium Ga 68-dotatate),用於檢測罕見的神經內分泌腫瘤(neuro-endocrine tumor, NETs)。NetspotTM是第一個鎵68-dotatate注射放射性診斷製劑,用於PET影像診斷與定位成人和兒童患者罕見的體抑素受體陽性神經內分泌腫瘤(somatostatin receptor positive neuroendocrine tumors)。NETs是一類罕見的良性或惡性腫瘤,多見於人體產生荷爾蒙的細胞中,而這些細胞遍布在胃部、小腸、胰腺與肺部等人體器官。這些腫瘤表面上,通常會有一種受體蛋白,負責結合生長激素抑制素(somatostatin),而鎵68-dotatate與somatostatin的結構接近,因此也能夠與這些受體結合,包括過度表現體抑素受體亞型2 (somatostatin receptor subtype 2, SSTR2)之癌細胞。由於鎵68-dotatate能夠發射正電子,並能被PET掃描所檢測,因此能夠精準地定位神經內分泌瘤。
2016年6月1日,美國FDA以孤兒藥地位和優先審查資格核准由總部位於法國Advanced Accelerator Application USA公司開發的一款新的診斷顯像劑NetspotTM(gallium Ga 68-dotatate),用於檢測罕見的神經內分泌腫瘤(neuro-endocrine tumor, NETs)。NetspotTM是第一個鎵68-dotatate注射放射性診斷製劑,用於PET影像診斷與定位成人和兒童患者罕見的體抑素受體陽性神經內分泌腫瘤(somatostatin receptor positive neuroendocrine tumors)。NETs是一類罕見的良性或惡性腫瘤,多見於人體產生荷爾蒙的細胞中,而這些細胞遍布在胃部、小腸、胰腺與肺部等人體器官。這些腫瘤表面上,通常會有一種受體蛋白,負責結合生長激素抑制素(somatostatin),而鎵68-dotatate與somatostatin的結構接近,因此也能夠與這些受體結合,包括過度表現體抑素受體亞型2 (somatostatin receptor subtype 2, SSTR2)之癌細胞。由於鎵68-dotatate能夠發射正電子,並能被PET掃描所檢測,因此能夠精準地定位神經內分泌瘤。FDA是基於3項臨床結果核准NetSpotTM:第一項研究是進行NETs患者以NetSpotTM或已獲准藥物在顯像診斷腫瘤定位之結果比較;第二項研究是利用組織病理學或臨床追蹤作為參考標準,對 NetSpotTM影像診斷進行評估;第三項研究是以NetSpotTM影像評估對NETs患者之復發情況。所有3項研究的結果證實NetSpotTM影像在發現NETs腫瘤定位之臨床有效性,且沒有發現嚴重副作用。
註1:新藥大放異彩-漫談2014年FDA核准小分子新藥(上),《國家衛生研究院電子報607期》;
新藥大放異彩-漫談2014年FDA核准小分子新藥(中),《國家衛生研究院電子報609期》;
新藥大放異彩-漫談2014年FDA核准小分子新藥(下),《國家衛生研究院電子報610期》。
註2:新藥再創新猷—漫談2015年美國FDA核准小分子新藥(上),《國家衛生研究院電子報659期》;
新藥再創新猷—漫談2015年美國FDA核准小分子新藥(中),《國家衛生研究院電子報660期》;
新藥再創新猷—漫談2015年美國FDA核准小分子新藥(下),《國家衛生研究院電子報661期》。
《文/圖:生技與藥物研究所謝興邦研究員、李靜琪副研究員》