NHRI Communications

知識產權
新藥回歸正常—漫談2016年FDA核准小分子新藥(中)
Introduction to FDA approved novel small molecular drugs in 2016 (part 2)
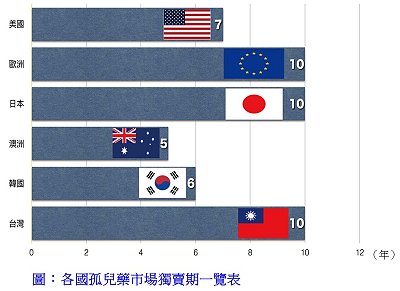 罕見疾病藥物「註1」
罕見疾病藥物「註1」延續2014年通過17個新藥(占當年所有通過藥物案件之41%)、2015年通過21個新藥(占當年所有通過藥物案件之47%),2016年則通過9個新藥(41%)是用於治療患者總數少於20萬人的罕見疾病用藥(47% - 2015年),其中包括1個生物藥和8個小分子藥物—Anthim®、Defitelio®、Exondys 51TM、Lartruvo®(單株抗體)、Netspot®、Ocaliva®、RubracaTM、Spinraza®、VenclextaTM。裡面有4個是針對癌症治療卅診斷—RubracaTM (癌症)、VenclextaTM (癌症)、Anthim®(放射性診斷劑)和Netspot®(放射性診斷劑),已於上期(711)專文報導[註2]。本期接續介紹另外4個罕見疾病藥物和2個外用藥物。
(1) Defitelio® (Defibrotide sodium)
2016年3月30日美國FDA以優先審查資格和孤兒藥地位核准總部設立於愛爾蘭都柏林市的Jazz Pharmaceuticals公司所開發的Defitelio®(defibrotide sodium),是第一個獲准用於治療成人和兒童因血液幹細胞移植(hematopoietic stem cell transplantation, hSCT)而造成的肝靜脈阻塞(veno-occlusive disease, VOD)藥物。肝靜脈阻塞是一種罕見卻致命的疾病,接受過高劑量化療以及血液幹細胞移植的血癌患者,有2%會引發嚴重肝靜脈栓塞,而其中高達80%的患者無法存活。肝靜脈阻塞的症狀為肝血管阻塞,進而導致肝內血液交換減少和腫脹,除了造成肝損傷,還有可能影響腎臟和肺功能。
Defitelio®的活性成分為單股多聚脫氧核苷酸(polydeoxyribonucleotide),在2016年以前,FDA共核准3個polydeoxyribonucleotide藥物,分別是Isis Pharmaceuticals / Novartis公司聯合開發用於治療巨細胞病毒(cytomegalovirus, CMV)性視網膜炎的Vitravene®、OSI Pharmaceuticals /EyeTech Pharmaceuticals / NeXstar Pharmaceuticals公司聯合開發用於治療新生血管性黃斑病變的Macugen®,以及Genzyme公司開發用於治療家族性高膽固醇血症(HoFH)的Kynamro®。值得一提的是,2016這一年,FDA破天荒一口氣通過3項polydeoxyribonucleotide藥物,除了Defitelio®外,還有首項用於治療裘馨氏肌肉萎縮症(Duchenne muscular dystrophy, DMD)的藥物Exondys 51 TM和首項用於治療脊髓性肌萎縮症(spinal muscular atrophy, SMA)兒童和成人藥物Spinraza®。
Defitelio®是一種具有纖溶酶特性的寡核苷酸混合物,萃取純化豬的腸粘膜而得,分子量約為13-21 kDa,能提高纖溶酶水解纖維蛋白凝塊的酶活性,增加組織纖溶酶原激活物t-PA和血栓調節蛋白表達,減少血管性血友病因子vWF和纖溶酶原激活物抑制劑PAI-1的表達。FDA是基於總數528位患者的3項臨床試驗結果(包括兩項前瞻性臨床試驗(prospective clinical trials )和1項延伸附加試驗(expanded access study))核准Defitelio®。3項研究的受試者均在血液幹細胞移植療程後確診出併發肝靜脈栓塞症,且伴隨有肝臟或腎臟異常。以Defitelio®治療的肝靜脈阻塞患者在血液幹細胞移植100天後的存活率為38 – 45%,而僅接受支持性療法的患者,其存活率只有21–31%。Defitelio®最常見的副作用包括:血壓異常低(低血壓)、腹瀉、嘔吐、噁心和流鼻血(鼻衄)。Defitelio®已確定的嚴重潛在副作用包括出血(hemorrhage)和過敏反應。目前在歐洲Defitelio® 的價格為33,000英鎊卅年(約四萬七千美元卅年),2015年在歐洲的銷售額為7,073萬美元,預計2016年在全球將會有1.05至1.20億美元的銷售額。
Defitelio®是由位於義大利Villa Guardia市的Gentium公司所研發的藥物,曾經在義大利上市,但由於商業原因而在2009年下市。2001年時,Gentium公司與位於美國馬里蘭州的Sigma-Tau Pharmaceuticals公司(母公司為Sigma-Tau Group,位於義大利Pomezia市)以50%:50%比例出資,用於Defitelio®在北美洲、中美洲和南美洲所有臨床試驗到藥證取得之費用,並由Sigma-Tau Pharmaceuticals公司取得上述地區之權利,Gentium公司則保有歐洲和上述以外的區域之權利,並可獲得總價值1,500萬美元之里程碑金以及上市後7%銷售權利金。之後,Gentium / Sigma-Tau Pharmaceuticals公司於2011年向美國提出新藥查驗登記(new drug application, NDA),結果36小時後就收到FDA的拒絕信。接著,他們轉戰歐洲,在2011年5月向歐洲藥物管理局(European Medicines Agency, EMA)提出NDA,經過1年審查,歐盟人用醫藥產品委員會(Committee for Medicinal Products for Human Use, CHMP)依然不同意核准,但經Gentium公司申請上訴,CHMP於是考量VOD現階段完全沒有任何治療藥物,故更改意見,於2013年10月18日,Defitelio®順利取得EMA核准上市。而後,Gentium公司在2013年12月20日被創立於2003年的Jazz Pharmaceuticals公司以10億美元收購,並取得Defitelio®歐洲和美洲以外之權利,隔年7月再以2億5千萬美元之里程碑金,從Sigma-Tau Pharmaceuticals公司手中買回Defitelio®在北美洲、中美洲和南美洲的權利。
(2) Exondys 51TM (Eteplirsen)
裘馨氏肌肉萎縮症是一種罕見的X染色體隱性遺傳疾病。據統計,全球平均每3,600個新生男嬰中就有一人罹患此疾病,這種疾病在沒有疾病家族史的群體中也時常發生。患者在兒童期就會因骨骼肌不斷退化出現肌肉無力或萎縮,導致行走不便。第一次症狀發生通常在3到5歲之間,並且逐漸惡化;大概到十幾歲時,會澈底喪失獨立活動能力而須使用輪椅;通常到20至30多歲,患者就會因發生心臟和呼吸系統疾病而死亡。2016年9月19日FDA以新藥快速審查資格、優先審查資格、孤兒藥地位和加速核准機制,有條件核准由位於麻薩諸塞州劍橋市專注於研發罕見傳染病生物藥的Sarepta Therapeutics公司所開發之Exondys 51TM(Eteplirsen; AVI-4658),是第一個治療DMD的藥物。Exondys 51TM為磷醯二胺嗎啉代反義寡核苷酸(antisense oligonucleotide, ASO),適用於肌縮蛋白(dystrophin)基因中存在確證突變可導致51號外顯子跳躍(exon 51 skipping)的DMD患者,此類患者約占 DMD 患者總數的13%。FDA有條件核准Exondys 51TM是基於12位DMD男孩患者接受Exondys 51TM治療後的結果,資料顯示於受試者骨骼肌中觀察到了肌縮蛋白的增加,故預測exon 51 skipping的DMD患者,可能會獲得臨床效益;因此要求Sarepta Therapeutics公司必須展開進一步的臨床研究,證明Exondys 51TM的確可以改善DMD患者活動力,始可將有條件核准轉變為完全核准。另外,由於DMD屬罕見兒科疾病,故Sarepta Therapeutics公司因為開發Exondys 51TM而獲得FDA給予FDA 優先審評憑證(priority review voucher),這是此制度實施以來第7個獲得兒童罕見病優先審評憑證的例子。
Exondys 51TM是由AVI BioPharma公司開發,該公司於2012年7月12日改名為Sarepta Therapeutics公司。Exondys 51TM是於2015年8月25日使用《The Prescription Drug User Fee Act(PDUFA)法案》提出NDA申請,故審查結果需於2016年2月26日前完成;後因FDA已先拒絕類似Exondys 51TM之治療DMD藥物 Drisapersen,故FDA提出3個月延長審查。FDA專家顧問委員會在2016年4月25日以3票贊成7票反對Exondys 51TM的申請,接著,FDA於2016年6月要求Sarepta Therapeutics公司提供更多的臨床證據來證明臨床試驗中患者肌縮蛋白表達恢復程度。在FDA收到額外附加的研究數據之後,內部意見依然分歧,因為12名患者中,僅有1人肌縮蛋白的表達有超過1%的增加,實在無法證明該藥物能夠帶來任何臨床效益。然而,FDA藥物評價與研究中心Janet Woodcock主任則駁回FDA專家顧問委員會決議,認為FDA應該核准Exondys 51TM,主要原因在於DMD是一種致命的兒童疾病,目前並沒有任何治療方法,因此建議FDA應該要靈活應變;最後在FDA Robert Califf局長的支持下,Exondys 51TM終於在2016年9月19日獲得FDA核准上市。目前估計使用Exondys 51TM一年費用約為三十萬美元,銷售高峰可達到一年10億美元。
(3) Spinraza® (Nusinersen)
脊髓性肌肉萎縮症為一個體染色體隱性遺傳疾病,目前已知造成此症的運動神經元存活(survival motor neuron, SMN)基因位於染色體 5q111.2 – 13.3這段區域,在這個位置上有兩個DNA序列非常相似的SMN基因:SMN1及SMN2。由於SMN1與SMN2基因序列區塊具有高度相似性,導致SMN1與SMN2基因容易發生缺失(deletion)或轉換(conversion)。約95%的SMA患者(包含第1、2與第3型)其SMN1基因發生缺失或轉換,其餘5%則屬於SMN1基因內的突變(intragenic mutation)。患者因先天的基因缺陷,導致脊髓前角運動神經細胞之衰亡與退化,使得肌肉逐漸無力、萎縮,身體逐漸喪失各種活動力,甚至是呼吸和吞嚥。SMA是2歲以下嬰幼兒群體中的頭號遺傳疾病殺手,是一種相對常見的「罕見病」,在新生兒中的患病率為1:6,000 – 1:10,000。
2016年12月23日FDA僅以3個月審查時間,快速地以新藥快速審查資格、優先審查資格和孤兒藥地位核准由Biogen與Ionis製藥公司所合作開發的Spinraza®(Nusinersen; IONIS-SMNRx;ISIS-SMNRx),是第一個治療SMA的藥物,更因此獲得FDA給予的FDA 優先審評憑證。Spinraza®也是一種反義寡核苷酸,用於5q脊髓性肌萎縮症(5q-SMA)的治療,這是SMA最常見的類型,約占全部SMA病例的95%。Spinraza®的作用在於改變SMN2 pre-mRNA的剪接(splicing),以增加全功能性SMN1蛋白的生產。FDA的核准是基於ENDEAR研究在嬰幼兒期發病型SMA(infantile-onset SMA,最有可能發展為第1型SMA)患者的結果。共有121位受試者,在6個月大之前確診患有SMA,在不到7個月大時開始第一次用藥。於此研究中,受試者分別接受 Spinraza®注射(注射入脊髓周圍的液體)或接受模擬程式(面板測試)作為對照。FDA要求研究團隊進行期中分析(interim analysis),以儘早審查試驗之有效性,結果共有82名SMA患者符合期中分析條件,有40%的Spinraza®治療患者顯示活動力指標改善,而對照組並未顯示有任何改善。
Spinraza®最早是由在美國冷泉港實驗室的Adrian Krainer博士和任職於Isis Pharmaceuticals公司(2015年12月改名為Ionis Pharmaceuticals公司)的資深副總裁Frank Bennett共同合作開發,於2011年12月19日進入人體臨床試驗。2012年1月4日Biogen公司和Isis Pharmaceuticals公司簽訂共同開發Spinraza®合約,Isis Pharmaceuticals公司獲得2,900萬美元簽約金以及4,500萬美元之不同臨床階段之里程碑金。當Spinraza®於2015年完成第一個臨床II卅III期試驗後,Biogen行使Spinraza®之技轉選擇權,Isis Pharmaceuticals公司因此獲得2.25億美元(包括7,500萬美元授權簽約金和1.5億美元藥品核准之里程碑金)以及15%產品上市之銷售權利金;同時,Biogen公司支付所有開發費用以及當初Isis Pharmaceuticals公司支付給冷泉港實驗室和麻州大學醫學院之技轉花費。Biogen預定將Spinraza®第一年價格訂為75萬美元,隨後每年的用藥成本為37.5萬美元,以美國有1.5萬名SMA患者來看,預估2025年的銷售額可能會達到17億美元。然而,由於75萬美元卅年的價格是罕見疾病藥物之天價,可能會影響支付者與保險業者嚴格審查哪些患者可以使用此藥物,並限制藥物使用之總量,如此一來,較輕微SMA患者可能會因此錯失藥物治療的機會。
(4) Ocaliva® (Obeticholic acid)
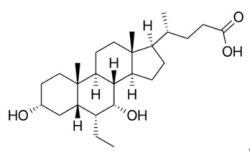 2016年5月27日FDA以新藥快速審查資格、孤兒藥地位和加速核准機制核准由位於紐約市的Intercept Pharmaceuticals公司所開發之Ocaliva®(Obeticholic acid;INT-747),可單獨使用或與ursodeoxycholic acid(UDCA)合併使用,用以治療針對UDCA不耐受的原發性膽汁性膽管炎(primary biliary cholangitis, PBC)成人患者。原發性膽汁性膽管炎是一種肝內膽小管漸進性損傷的慢性疾病,主要是因膽管遭到自身免疫性破壞,導致膽汁淤積,主要影響女性,目前是美國女性進行肝臟移植手術的第二大病因;在歐洲,該病約占膽汁淤積性疾病所致肝移植病例的一半左右,約占所有肝移植病例的6%。UDCA是目前唯一獲核准用於治療原發性膽汁性膽管炎的藥物,可有效治療超過50%的患者,但多達40%的患者的ALP水平或總膽紅素不會降低,且5%–10%的患者無法耐受UDCA。 Ocaliva®成為近20年來核准治療原發性膽汁性膽管炎的首個新藥。
2016年5月27日FDA以新藥快速審查資格、孤兒藥地位和加速核准機制核准由位於紐約市的Intercept Pharmaceuticals公司所開發之Ocaliva®(Obeticholic acid;INT-747),可單獨使用或與ursodeoxycholic acid(UDCA)合併使用,用以治療針對UDCA不耐受的原發性膽汁性膽管炎(primary biliary cholangitis, PBC)成人患者。原發性膽汁性膽管炎是一種肝內膽小管漸進性損傷的慢性疾病,主要是因膽管遭到自身免疫性破壞,導致膽汁淤積,主要影響女性,目前是美國女性進行肝臟移植手術的第二大病因;在歐洲,該病約占膽汁淤積性疾病所致肝移植病例的一半左右,約占所有肝移植病例的6%。UDCA是目前唯一獲核准用於治療原發性膽汁性膽管炎的藥物,可有效治療超過50%的患者,但多達40%的患者的ALP水平或總膽紅素不會降低,且5%–10%的患者無法耐受UDCA。 Ocaliva®成為近20年來核准治療原發性膽汁性膽管炎的首個新藥。繼2015年通過二個膽酸(bile acid)衍生物—治療消除雙下巴藥物Kybella®(deoxycholic acid)和治療膽汁酸合成障礙藥物Cholbam®(cholic acid),Ocaliva®也是一個半合成的膽酸衍生物,其作用機制為法尼醇X受體(farnesoid X receptor, FXR )促效劑,促進膽汁酸的釋放,可用於原發性膽汁性膽管炎、非酒精性脂肪性肝炎(non-alcoholic steatohepatitis, NASH)、原發性硬化性膽管炎(primary sclerosing cholangitis, PSC)及其他肝臟疾病和腸道疾病的治療。FDA是基於包括有216位原發性膽汁性膽管炎患者的臨床試驗結果核准Ocaliva®。相對於對照組,以Ocaliva®治療1年後的患者的鹼性磷酸酶ALP水平降低。Ocaliva®是在2001年由義大利University of Perugia 的Roberto Pellicciari教授發現,並於2002年9月4日與 Mark E. Pruzanski醫師一起創辦Intercept Pharmaceuticals公司開發此藥物。接著於2011年3月30日與日本Dainippon Sumitomo Pharma公司簽訂技轉合約,由Dainippon Sumitomo Pharma公司取得日本、中國、韓國和台灣的Ocaliva®所有權,而Intercept Pharmaceuticals公司從Dainippon Sumitomo Pharma公司取得1,500萬美元簽約金、3億美元的里程碑金以及10%上述授權區域之銷售權利金。目前Ocaliva®的定價為1年7萬美元,預估2020年可達到16億美元之銷售額。原發性膽汁性膽管炎是Ocaliva®獲核准的首個適應症,目前Ocaliva®仍在開發用於多種慢性肝臟疾病的治療,包括NASH(FDA已授予突破性藥物資格)、PSC和膽道閉鎖,這可能會是價值數十億美元的市場,故EvaluatePharma公司預測Ocaliva®在2020年的銷售額將高達30億美元。
外用藥物
(1) XiidraTM
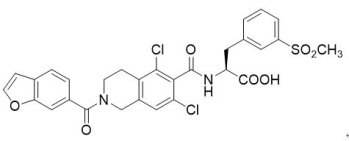 乾眼症(dry eye disease, DED)又名角結膜乾燥症(keratoconjunctivitis sicca, KCS),是現代人極為常見的疾病。眼睛易紅腫疼痛,且容易受感染,所引發的炎症有可能導致眼球表面損傷;主要成因有二大類:一類是淚液分泌不足,另一類是淚液蒸發量過高。淚液分泌不足是最常見的乾眼原因,例如睡眠不足、老化、自體免疫疾病,甚至像外傷、感染、自律神經失調、長期使用青光眼藥物或服用高血壓藥或鎮靜劑、或長期戴隱形眼鏡等,都會造成淚液分泌不足。淚液蒸發量過高則是近年來漸受重視的乾眼症類型,成因如油脂層分泌不足、黏液素層分泌不足、缺乏維生素A、慢性結膜炎、類天皰瘡、化學性灼傷等,另外長時間使用電腦或手機以致眨眼次數減少、長時間在冷氣房或戶外強風乾燥之工作環境,甚至是眼瞼疾病造成眼瞼閉合不良等,都是可能造成乾眼症的因素,傳統治療方法是使用人工淚液。
乾眼症(dry eye disease, DED)又名角結膜乾燥症(keratoconjunctivitis sicca, KCS),是現代人極為常見的疾病。眼睛易紅腫疼痛,且容易受感染,所引發的炎症有可能導致眼球表面損傷;主要成因有二大類:一類是淚液分泌不足,另一類是淚液蒸發量過高。淚液分泌不足是最常見的乾眼原因,例如睡眠不足、老化、自體免疫疾病,甚至像外傷、感染、自律神經失調、長期使用青光眼藥物或服用高血壓藥或鎮靜劑、或長期戴隱形眼鏡等,都會造成淚液分泌不足。淚液蒸發量過高則是近年來漸受重視的乾眼症類型,成因如油脂層分泌不足、黏液素層分泌不足、缺乏維生素A、慢性結膜炎、類天皰瘡、化學性灼傷等,另外長時間使用電腦或手機以致眨眼次數減少、長時間在冷氣房或戶外強風乾燥之工作環境,甚至是眼瞼疾病造成眼瞼閉合不良等,都是可能造成乾眼症的因素,傳統治療方法是使用人工淚液。2016年7月12日,FDA以優先審查資格核准由總部在愛爾蘭都柏林市、而美國分公司位於麻州Lexington市的Shire US公司所開發的XiidraTM滴眼劑溶液(5%濃度的lifitegrast眼用溶液,SAR 1118),用以治療成人乾眼症患者,每日使用兩次。最近研究發現,乾眼症是一種發炎過程,而發炎可能最終導致眼睛表面損傷及相關聯的慢性眼部疾病,故乾眼症有關的發炎被認為是由T細胞和相關的細胞因子主要介導,當ICAM-1在角膜和結膜組織中過量表達時,LFA-1/ICAM-1的相互作用會促進免疫突觸的形成,導致T細胞活化並遷移至目標組織,而XiidraTM為LFA-1拮抗劑,可以阻斷LFA-1與ICAM-1的相互作用,進而防止發炎發生,以治療乾眼症。
XiidraTM的安全性與有效性評估是基於包含一千多位年紀介於19 – 97歲的患者,其中76%為女性的臨床試驗結果。隨機給予患者XiidraTM或安慰劑,每天使用兩次,療程持續12週;結果發現,接受XiidraTM眼藥水的患者,比用安慰劑的患者,症狀顯著改善。更重要的是,兩週即有療效。XiidraTM是過去13年來首個核准治療乾眼症的新藥,並且是唯一一款可同時改善乾眼症症狀和體徵的處方滴眼劑。XiidraTM是在2006年由3位原任職於Genentech公司的同事:Tom Gadek、John Bernier和Chuck O’Neill共同創立的SARcode Bioscience新創公司所研發,此公司在2013年3月25日被Shire公司以1.6億美元併購,當時XiidraTM正進行治療乾眼症之臨床III期試驗。
乾眼症的發生率,在30到40歲的族群大約是5%;在65歲以上的族群則大約是10% 到15%。據估計,全球大約有6,000萬乾眼症患者,在美國有1,600萬成年人被診斷患有乾眼症,因此業界對XiidraTM商業前景十分看好,預計年銷售額峰值將突破20億美元,同時也將成為Allergan乾眼症藥物Restasis®(cyclosporine-A (CsA) 0.05% ophthalmic emulsion)的強力競爭對手。Restasis®是一種免疫抑制劑,定位為一種人工淚液的替代品,唯一功效是增加淚水,而非改善症狀。儘管2種藥物尚未有一對一的比較研究,但臨床數據顯示,XiidraTM大約2週就能緩解病情,而Restasis®則需要6週或更長時間。在價格方面,XiidraTM售價與Restasis®一致,一年大約需五千美元。根據Allergan的財務報告,Restasis®在2015年銷售額大約為十三億美元,儘管XiidraTM將對Restasis®帶來嚴重威脅,但市場上2種產品的激烈對抗所伴隨而來對乾眼症治療意識的提高,實際上同樣也會使Restasis®受益。
(2) Eucrisa® (Crisaborole)
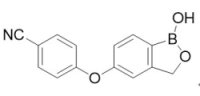 2016年12月14日,FDA以優先審查資格提前2個月核准由Pfizer藥廠在2016年6月以52億美元併購位於美國加州的Anacor藥廠所開發的Eucrisa®軟膏(2% Crisaborole;AN2728;PF-06930164),用以治療輕度至中度濕疹(eczema)(異位性皮膚炎,atopic dermatitis),適用於2歲以上的患者。異位性皮膚炎是一種慢性發炎皮膚病,被稱為「濕疹」,通常在兒童期開始發作,可以持續至成年,其致病原因是遺傳、免疫和環境因素的組合。Eucrisa®是一種含硼的小分子抗發炎局部外用藥物,每日兩次,是磷酸雙酯酶-4B(PDE-4B)選擇性抑制劑。PDE-4B與發炎相關,抑制PDE-4B活性,可有效抑制TNFα、 IL-12、 IL-23等之生成。
2016年12月14日,FDA以優先審查資格提前2個月核准由Pfizer藥廠在2016年6月以52億美元併購位於美國加州的Anacor藥廠所開發的Eucrisa®軟膏(2% Crisaborole;AN2728;PF-06930164),用以治療輕度至中度濕疹(eczema)(異位性皮膚炎,atopic dermatitis),適用於2歲以上的患者。異位性皮膚炎是一種慢性發炎皮膚病,被稱為「濕疹」,通常在兒童期開始發作,可以持續至成年,其致病原因是遺傳、免疫和環境因素的組合。Eucrisa®是一種含硼的小分子抗發炎局部外用藥物,每日兩次,是磷酸雙酯酶-4B(PDE-4B)選擇性抑制劑。PDE-4B與發炎相關,抑制PDE-4B活性,可有效抑制TNFα、 IL-12、 IL-23等之生成。FDA是基於二個臨床III期試驗結果確立Eucrisa®的安全性和功效,共有1,522名2歲到79歲具有輕度至中度異位性皮膚炎的患者參與臨床研究;相較於安慰劑組,使用Eucrisa®的患者在經過 28天治療後,病情顯著獲得緩解,濕疹完全消退或接近完全消退。
據估計,美國大約有二千五百萬人受到異位性皮膚炎之困擾,其中嬰兒和兒童占8 ~ 18%。目前異位性皮膚炎尚缺少有效的治療藥物,Eucrisa®的核准上市是近15年來FDA所通過的第一個用於治療異位性皮膚炎的非類固醇抗發炎藥物(non-steroidal anti-inflmmatory drug, NSAIDs)替代療法。業界對Eucrisa®商業前景十分看好,預計年銷售額峰值將突破20億美元。
[註1]新藥投資策略:孤兒藥篇,羅敏菁,2016 / 08 / 01。
[註2]新藥回歸正常—漫談2016年FDA核准小分子新藥(上),《國家衛生研究院電子報711期》。
《文/圖:生技與藥物研究所謝興邦研究員、李靜琪副研究員》